

Quantitative single-cell proteomics as a tool to characterize cellular hierarchies
- PMID: 34099695
- PMCID: PMC8185083
- DOI: 10.1038/s41467-021-23667-y
Quantitative single-cell proteomics as a tool to characterize cellular hierarchies
Abstract
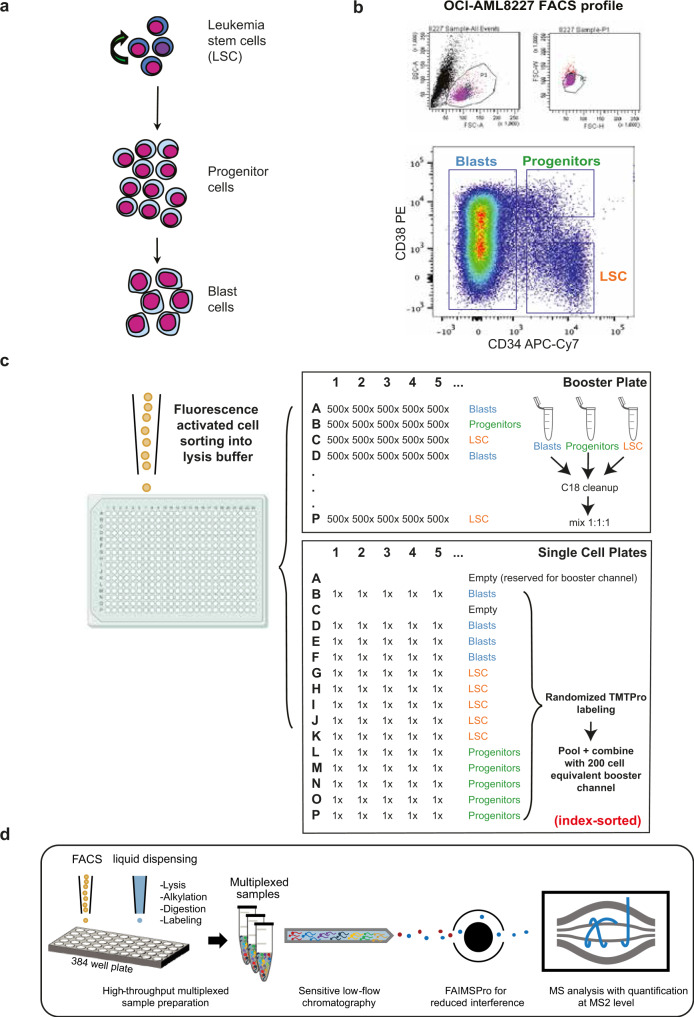
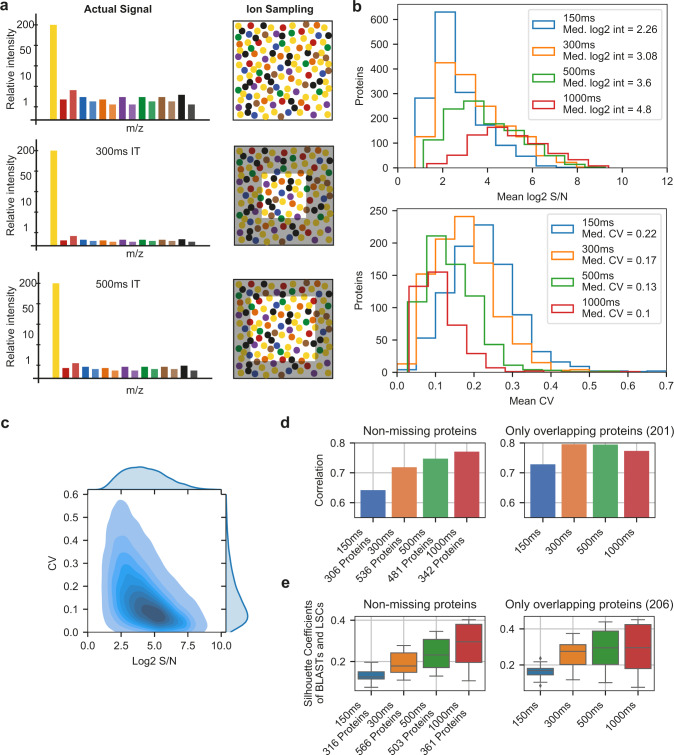
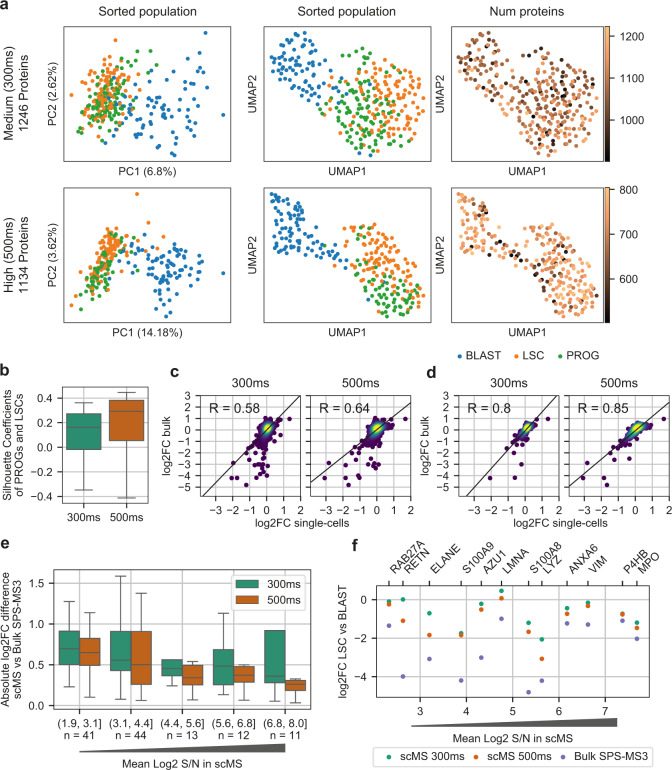
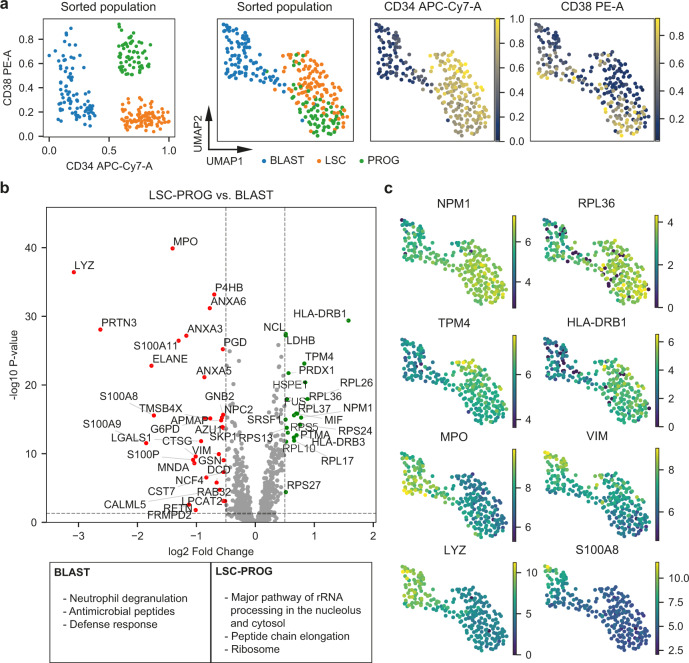
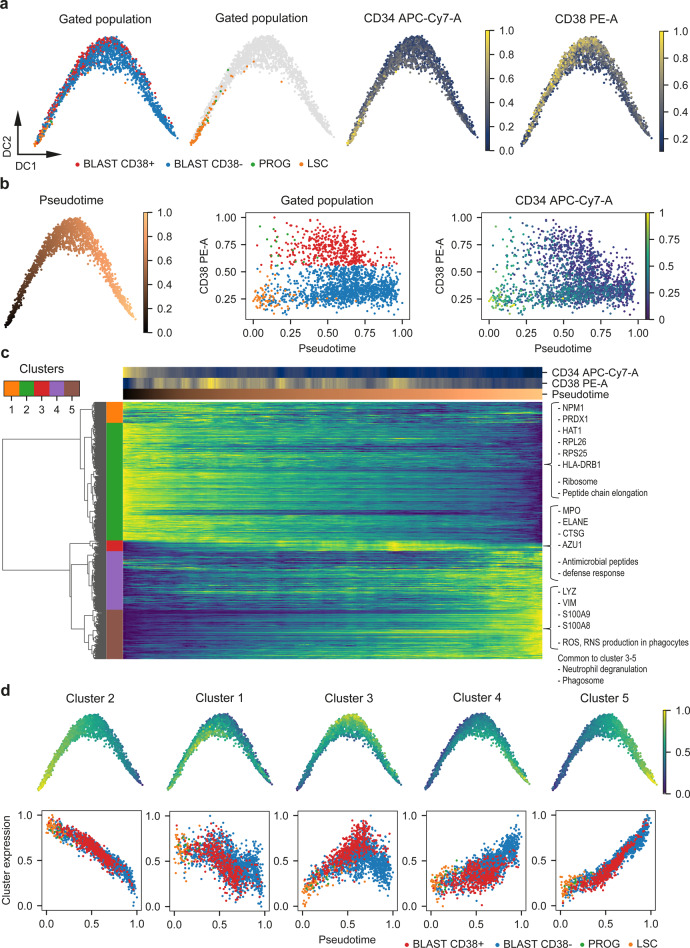
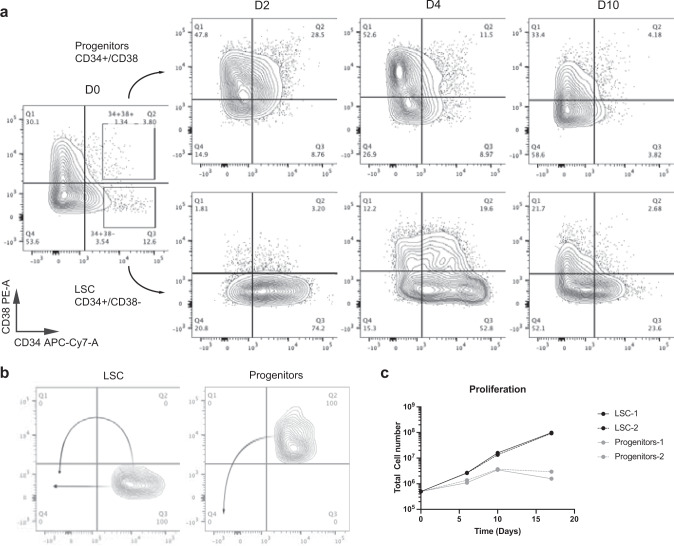
Large-scale single-cell analyses are of fundamental importance in order to capture biological heterogeneity within complex cell systems, but have largely been limited to RNA-based technologies. Here we present a comprehensive benchmarked experimental and computational workflow, which establishes global single-cell mass spectrometry-based proteomics as a tool for large-scale single-cell analyses. By exploiting a primary leukemia model system, we demonstrate both through pre-enrichment of cell populations and through a non-enriched unbiased approach that our workflow enables the exploration of cellular heterogeneity within this aberrant developmental hierarchy. Our approach is capable of consistently quantifying ~1000 proteins per cell across thousands of individual cells using limited instrument time. Furthermore, we develop a computational workflow (SCeptre) that effectively normalizes the data, integrates available FACS data and facilitates downstream analysis. The approach presented here lays a foundation for implementing global single-cell proteomics studies across the world.
Conflict of interest statement
The authors declare no competing interests.
Figures

Comment in
-
Towards resolving proteomes in single cells.Nat Methods. 2021 Aug;18(8):856. doi: 10.1038/s41592-021-01243-y. Nat Methods. 2021. PMID: 34354289 No abstract available.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
