

Live imaging and biophysical modeling support a button-based mechanism of somatic homolog pairing in Drosophila
- PMID: 34100718
- PMCID: PMC8294847
- DOI: 10.7554/eLife.64412
Live imaging and biophysical modeling support a button-based mechanism of somatic homolog pairing in Drosophila
Abstract
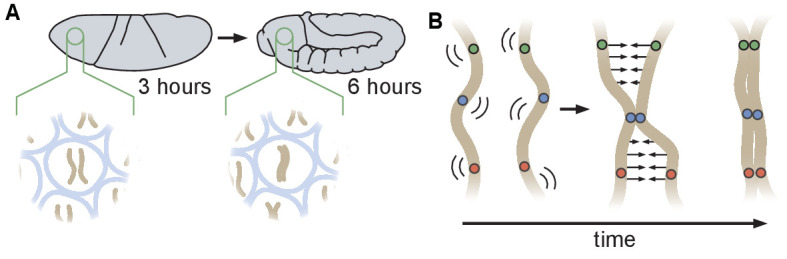
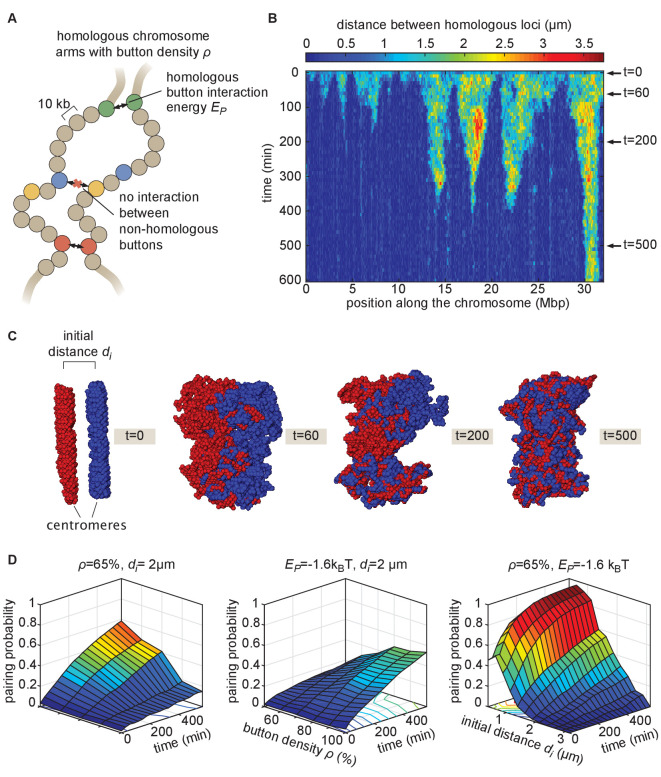
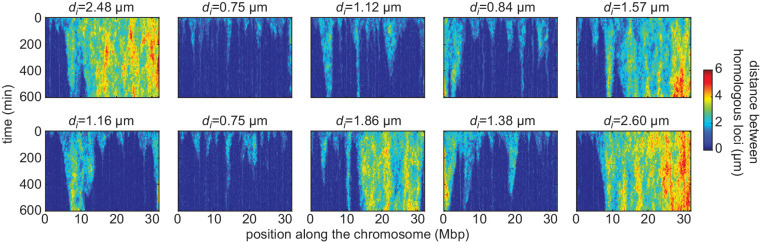
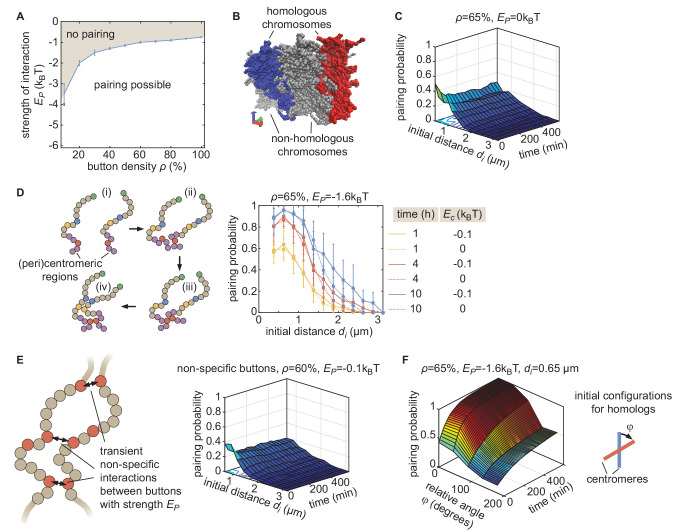
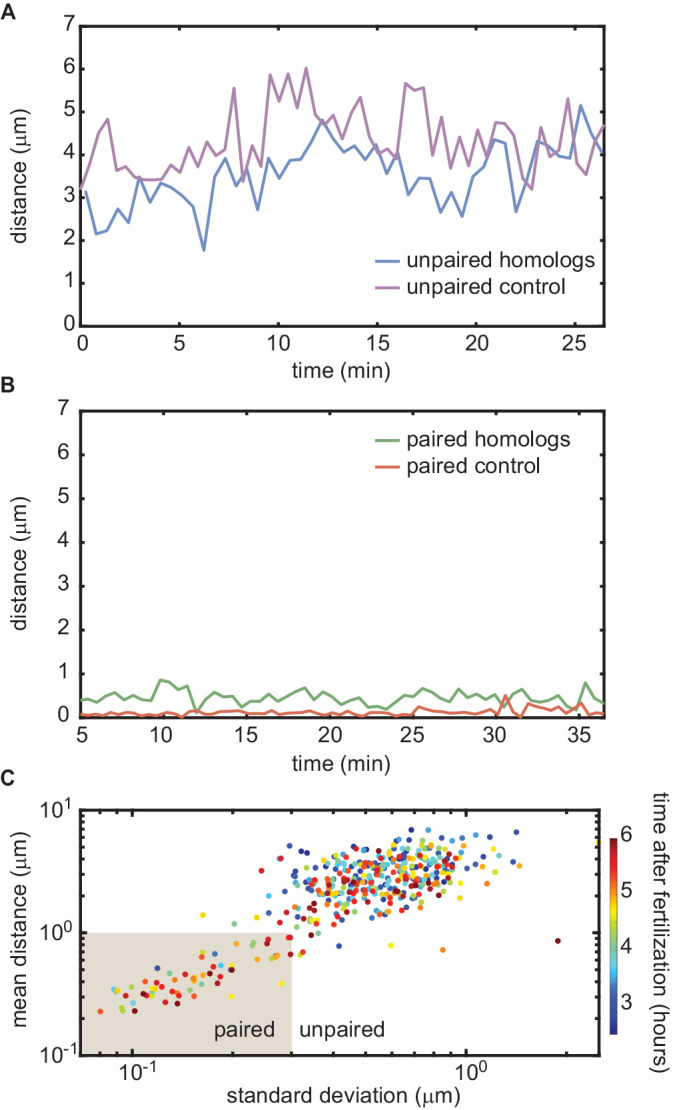
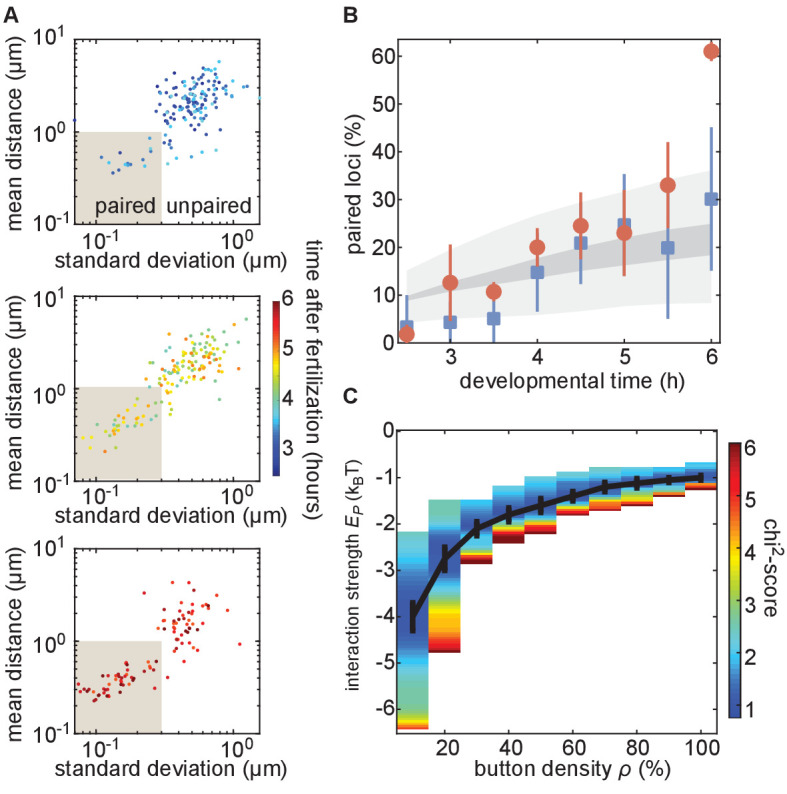
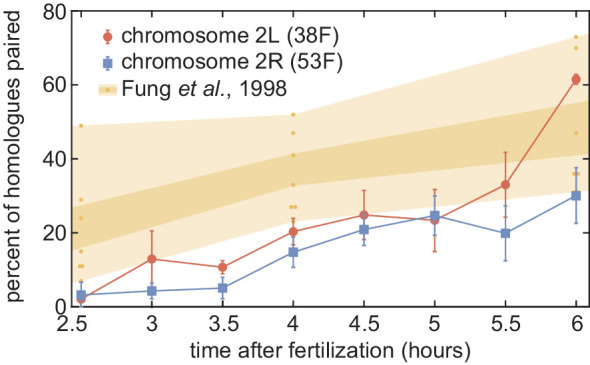
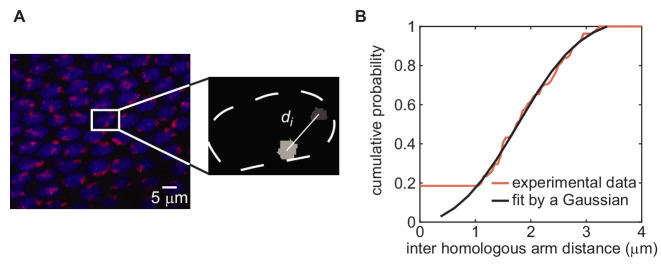
Three-dimensional eukaryotic genome organization provides the structural basis for gene regulation. In Drosophila melanogaster, genome folding is characterized by somatic homolog pairing, where homologous chromosomes are intimately paired from end to end; however, how homologs identify one another and pair has remained mysterious. Recently, this process has been proposed to be driven by specifically interacting 'buttons' encoded along chromosomes. Here, we turned this hypothesis into a quantitative biophysical model to demonstrate that a button-based mechanism can lead to chromosome-wide pairing. We tested our model using live-imaging measurements of chromosomal loci tagged with the MS2 and PP7 nascent RNA labeling systems. We show solid agreement between model predictions and experiments in the pairing dynamics of individual homologous loci. Our results strongly support a button-based mechanism of somatic homolog pairing in Drosophila and provide a theoretical framework for revealing the molecular identity and regulation of buttons.
Keywords: D. melanogaster; Drosophila; button model; homolog; homologous chromosomes; live imaging; pairing; physics of living systems.
© 2021, Child et al.
Conflict of interest statement
MC, JB, AJ, AR, NL, NS, DV, GM, JJ, DJ, HG No competing interests declared
Figures
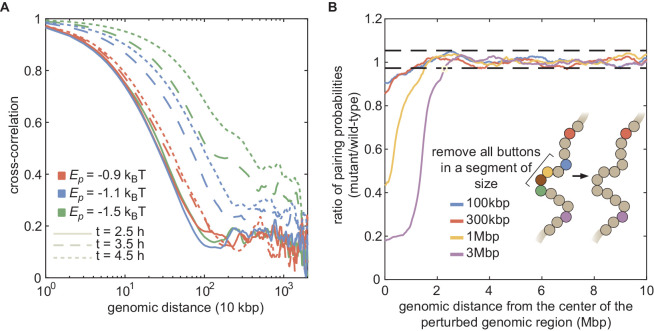
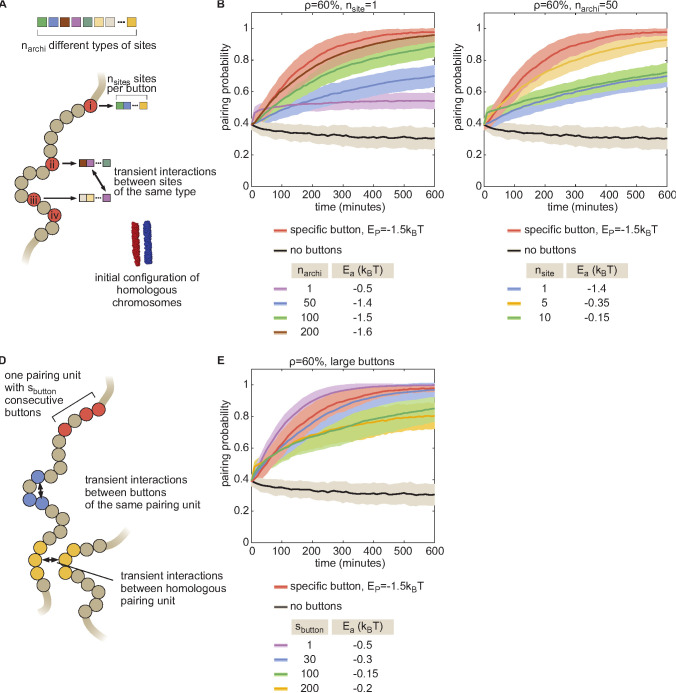
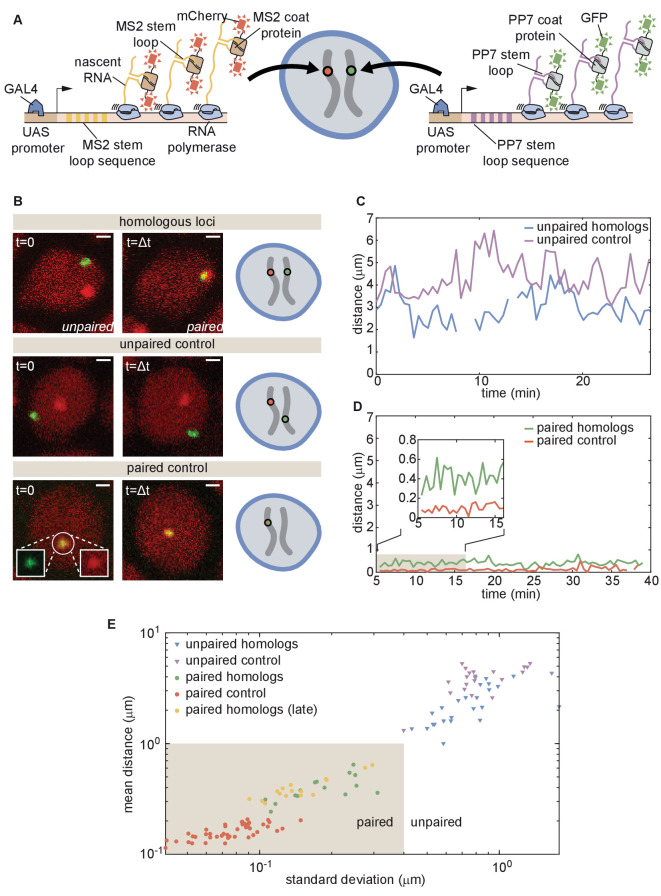
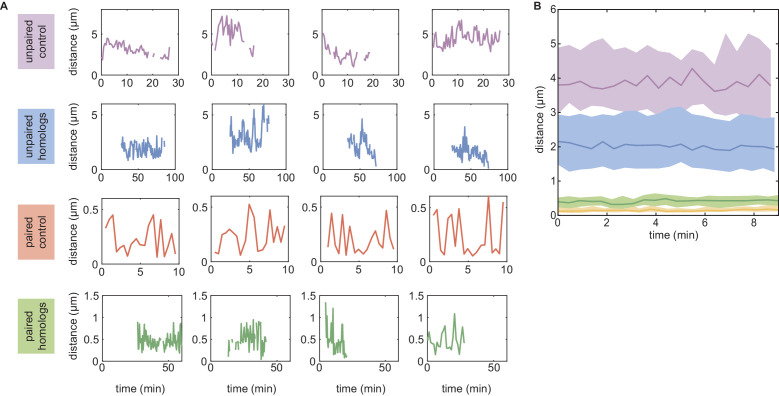
References
-
- AlHaj Abed J, Erceg J, Goloborodko A, Nguyen SC, McCole RB, Saylor W, Fudenberg G, Lajoie BR, Dekker J, Mirny LA, Wu CT. Highly structured homolog pairing reflects functional organization of the Drosophila genome. Nature Communications. 2019;10:4485. doi: 10.1038/s41467-019-12208-3. - DOI - PMC - PubMed
Publication types
MeSH terms
Associated data
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
