

Age-dependent neurological phenotypes in a mouse model of PRRT2-related diseases
- PMID: 34101060
- PMCID: PMC8241743
- DOI: 10.1007/s10048-021-00645-6
Age-dependent neurological phenotypes in a mouse model of PRRT2-related diseases
Abstract
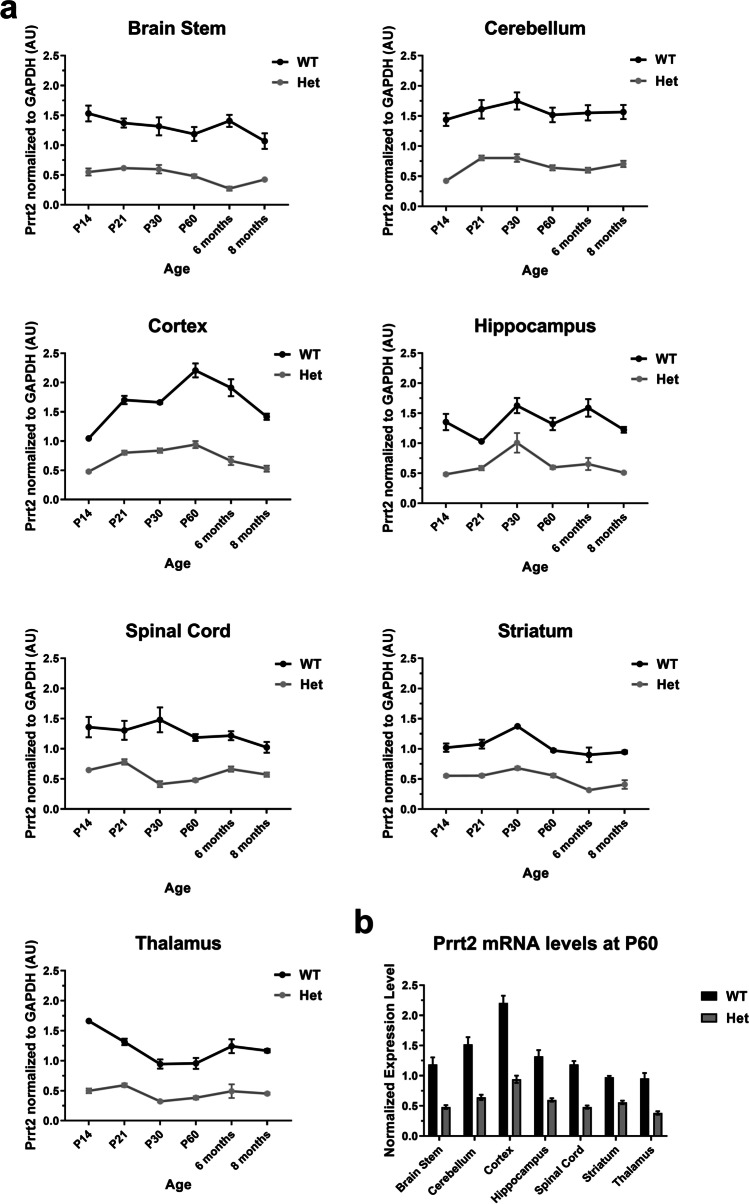
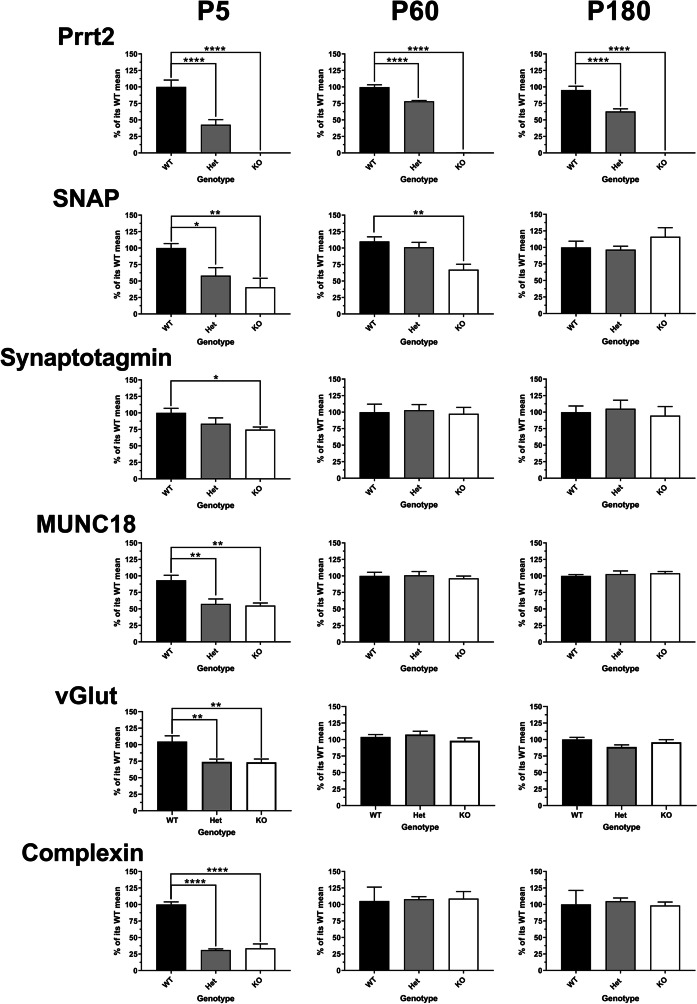
Paroxysmal kinesigenic dyskinesia is an episodic movement disorder caused by dominant mutations in the proline-rich transmembrane protein PRRT2, with onset in childhood and typically with improvement or resolution by middle age. Mutations in the same gene may also cause benign infantile seizures, which begin in the first year of life and typically remit by the age of 2 years. Many details of PRRT2 function at the synapse, and the effects of mutations on neuronal excitability in the pathophysiology of epilepsy and dyskinesia, have emerged through the work of several groups over the last decade. However, the age dependence of the phenotypes has not been explored in detail in transgenic models. Here, we report our findings in heterozygous and homozygous Prrt2 knockout mice that recapitulate the age dependence of dyskinesia seen in the human disease. We show that Prrt2 deletion reduces the levels of synaptic proteins in a dose-dependent manner that is most pronounced at postnatal day 5 (P5), attenuates at P60, and disappears by P180. In a test for foot slippage while crossing a balance beam, transient loss of coordination was most pronounced at P60 and less prominent at age extremes. Slower traverse time was noted in homozygous knockout mice only, consistent with the ataxia seen in rare individuals with biallelic loss of function mutations in Prrt2. We thus identify three age-dependent phenotypic windows in the mouse model, which recapitulate the pattern seen in humans with PRRT2-related diseases.
Keywords: Dyskinesia; Epilepsy; Mouse model; Movement disorders; Paroxysmal disorders.
Conflict of interest statement
The authors declare no competing interests.
Figures






References
-
- Wang JL, Cao L, Li XH, Hu ZM, Li JD, Zhang JG, Liang Y, San A, Li N, Chen SQ, Guo JF, Jiang H, Shen L, Zheng L, Mao X, Yan WQ, Zhou Y, Shi YT, Ai SX, Dai MZ, Zhang P, Xia K, Chen SD, Tang BS. Identification of PRRT2 as the causative gene of paroxysmal kinesigenic dyskinesias. Brain. 2021;134(Pt 12):3493–3501. doi: 10.1093/brain/awr289. - DOI - PMC - PubMed
-
- Chen WJ, Lin Y, Xiong ZQ, Wei W, Ni W, Tan GH, Guo SL, He J, Chen YF, Zhang QJ, Li HF, Lin Y, Murong SX, Xu J, Wang N, Wu ZY. Exome sequencing identifies truncating mutations in PRRT2 that cause paroxysmal kinesigenic dyskinesia. Nat Genet. 2011;43(12):1252–1255. doi: 10.1038/ng.1008. - DOI - PubMed
-
- van Vliet R, Breedveld G, de Rijk-van AJ, Brilstra E, Verbeek N, Verschuuren-Bemelmans C, Boon M, Samijn J, Diderich K, van de Laar I, Oostra B, Bonifati V, Maat-Kievit A. PRRT2 phenotypes and penetrance of paroxysmal kinesigenic dyskinesia and infantile convulsions. Neurology. 2012;79(8):777–784. doi: 10.1212/WNL.0b013e3182661fe3. - DOI - PubMed
-
- Cloarec R, Bruneau N, Rudolf G, Massacrier A, Salmi M, Bataillard M, Boulay C, Caraballo R, Fejerman N, Genton P, Hirsch E, Hunter A, Lesca G, Motte J, Roubertie A, Sanlaville D, Wong SW, Fu YH, Rochette J, Ptácek LJ, Szepetowski P. PRRT2 links infantile convulsions and paroxysmal dyskinesia with migraine. Neurology. 2012;79(21):2097–2103. doi: 10.1212/WNL.0b013e3182752c46. - DOI - PMC - PubMed
-
- Lee HY, Huang Y, Bruneau N, Roll P, Roberson ED, Hermann M, Quinn E, Maas J, Edwards R, Ashizawa T, Baykan B, Bhatia K, Bressman S, Bruno MK, Brunt ER, Caraballo R, Echenne R, Fejerman N, Frucht S, Gurnett CA, Hirsch E, Houlden H, Jankovic J, Lee WL, Lynch DR, Mohammed S, Müller U, Nespeca MP, Renner D, Rochette J, Rudolf G, Saiki S, Soong BW, Swoboda KJ, Tucker S, Wood N, Hanna M, Bowcock AM, Szepetowski P, Fu YH, Ptáček LJ. Mutations in the gene PRRT2 cause paroxysmal kinesigenic dyskinesia with infantile convulsions. Cell Rep. 2012;1(1):2–12. doi: 10.1016/j.celrep.2011.11.001. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Miscellaneous
