

Inhibition of Nav1.7 channel by a novel blocker QLS-81 for alleviation of neuropathic pain
- PMID: 34103689
- PMCID: PMC8285378
- DOI: 10.1038/s41401-021-00682-9
Inhibition of Nav1.7 channel by a novel blocker QLS-81 for alleviation of neuropathic pain
Abstract
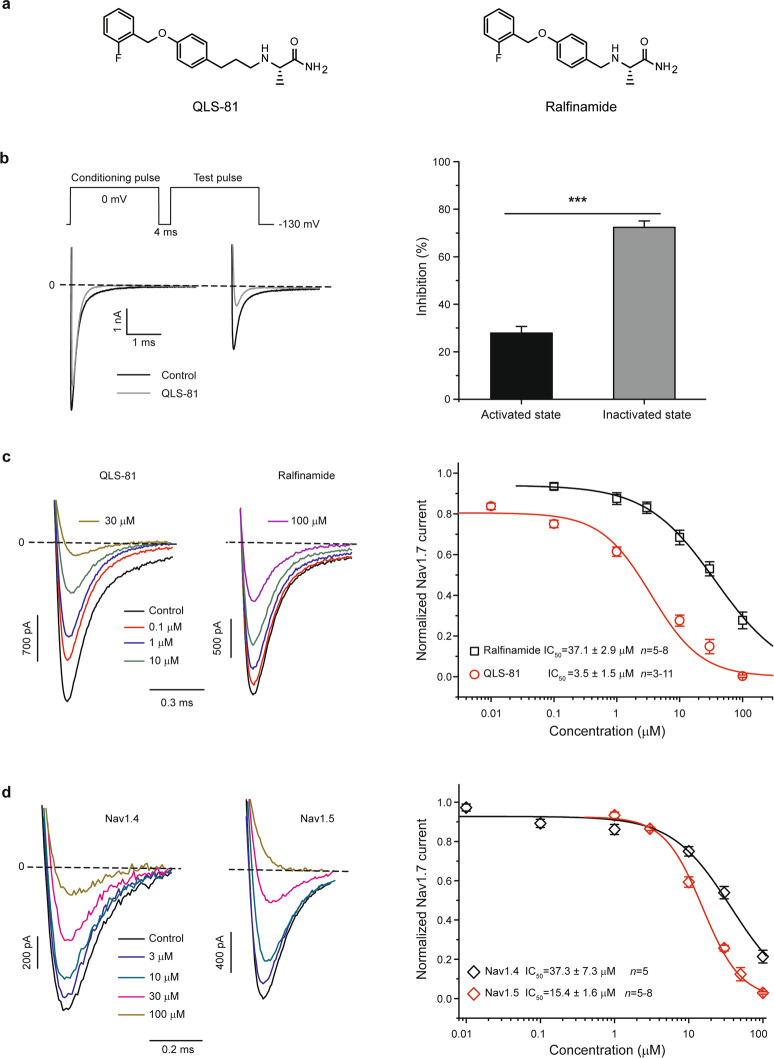
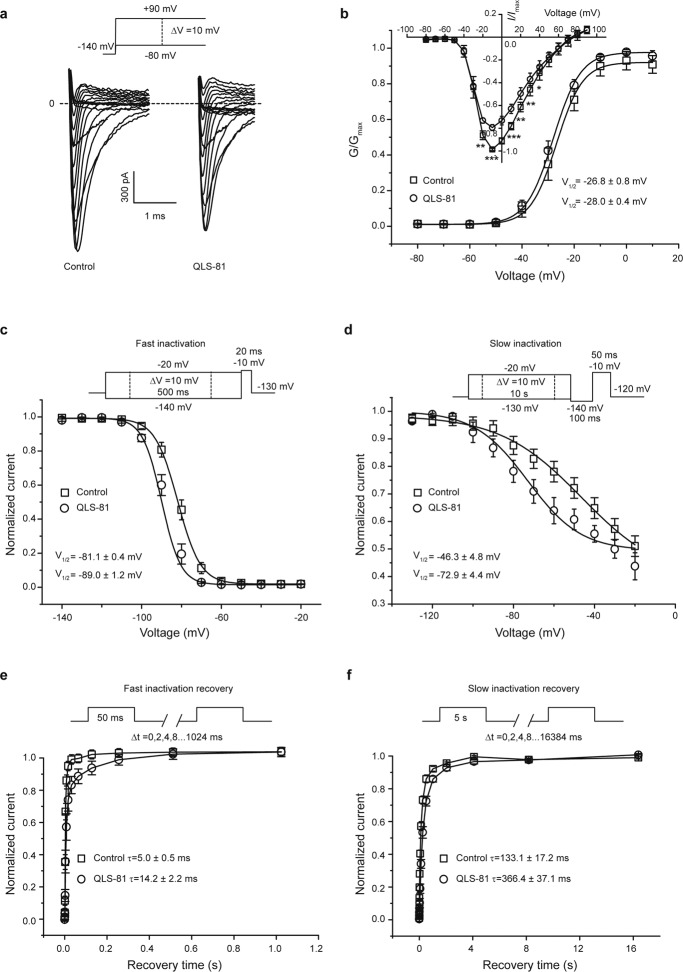
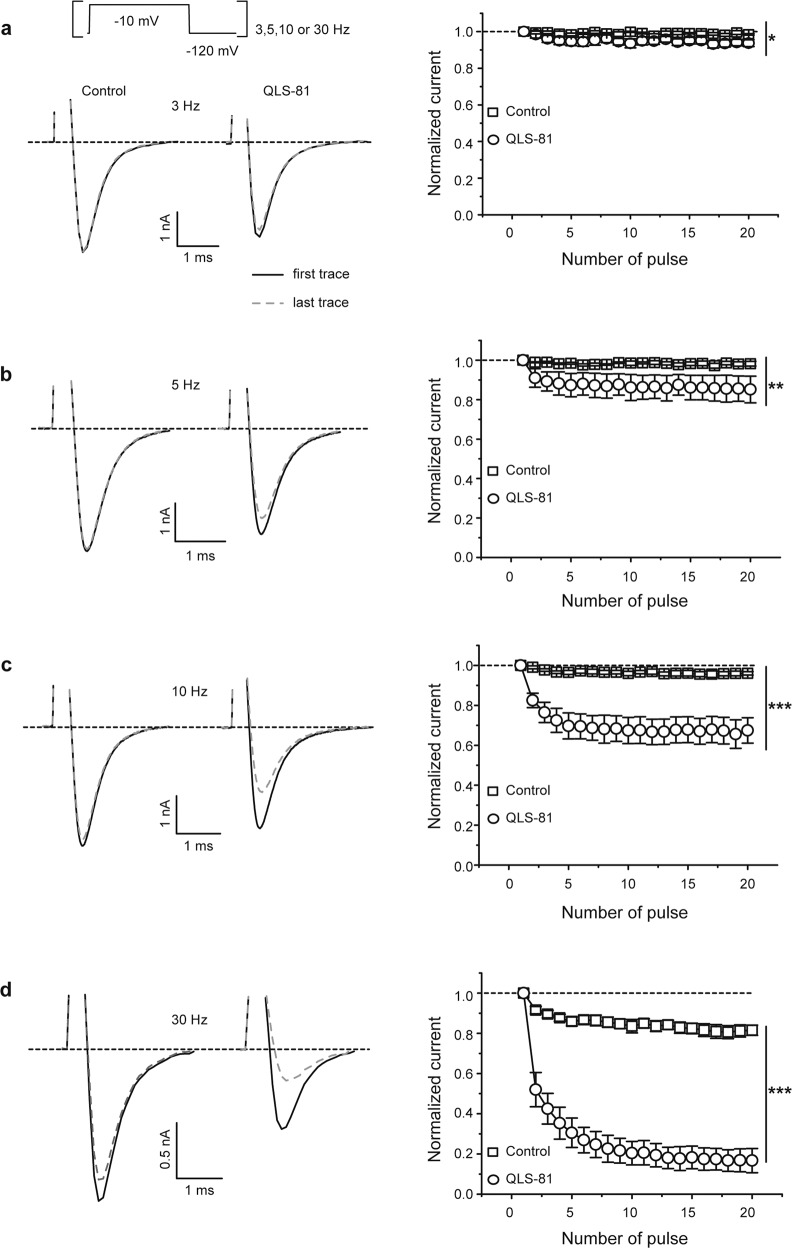
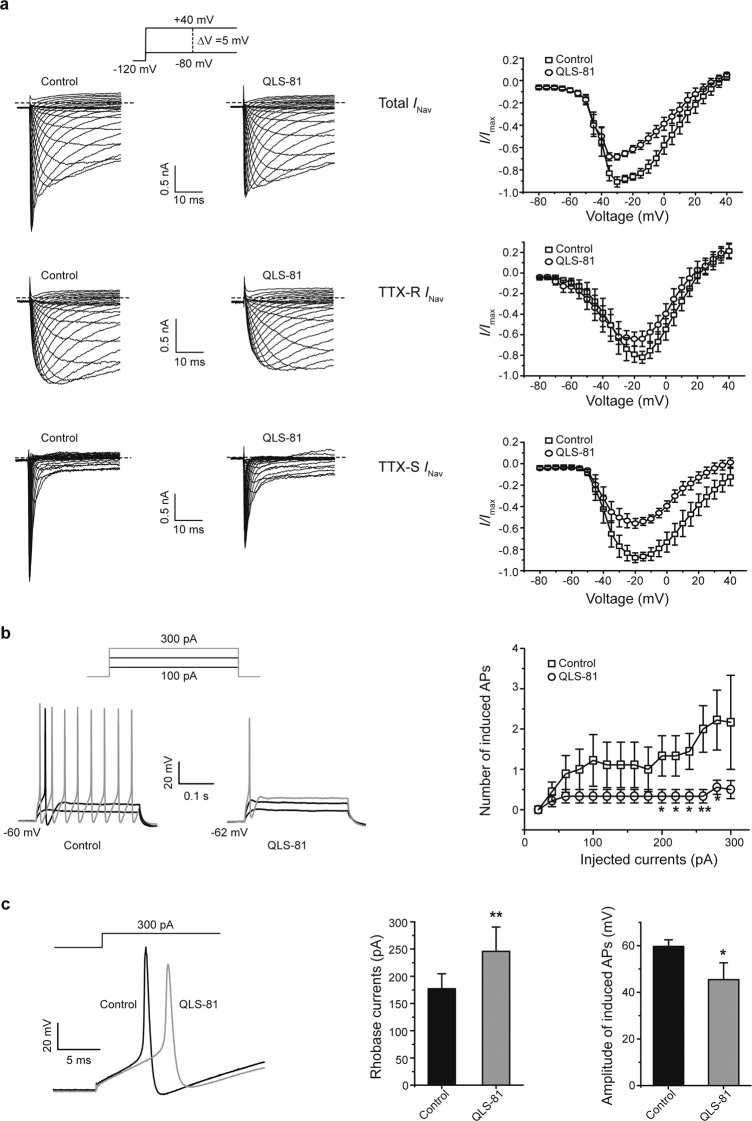
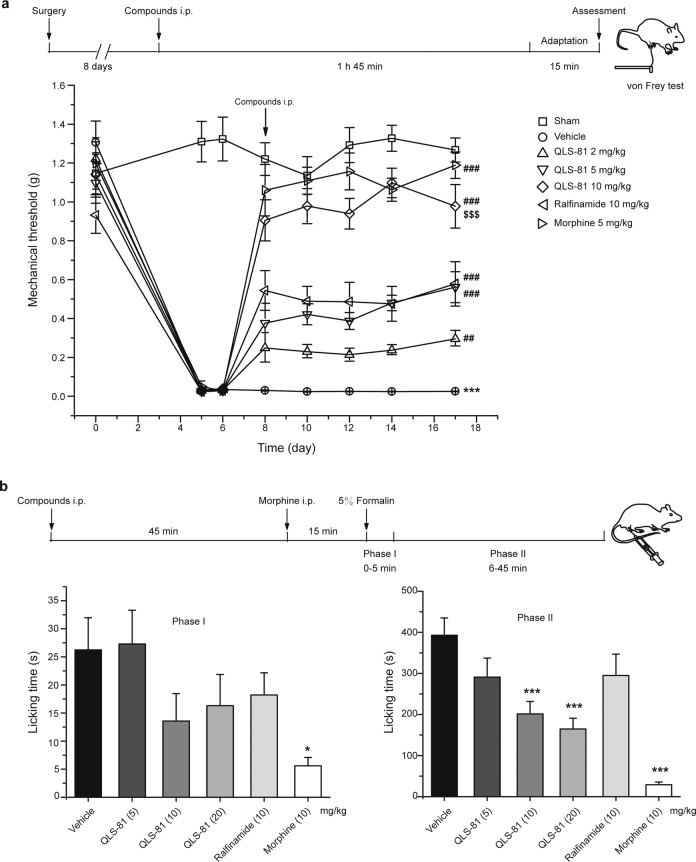
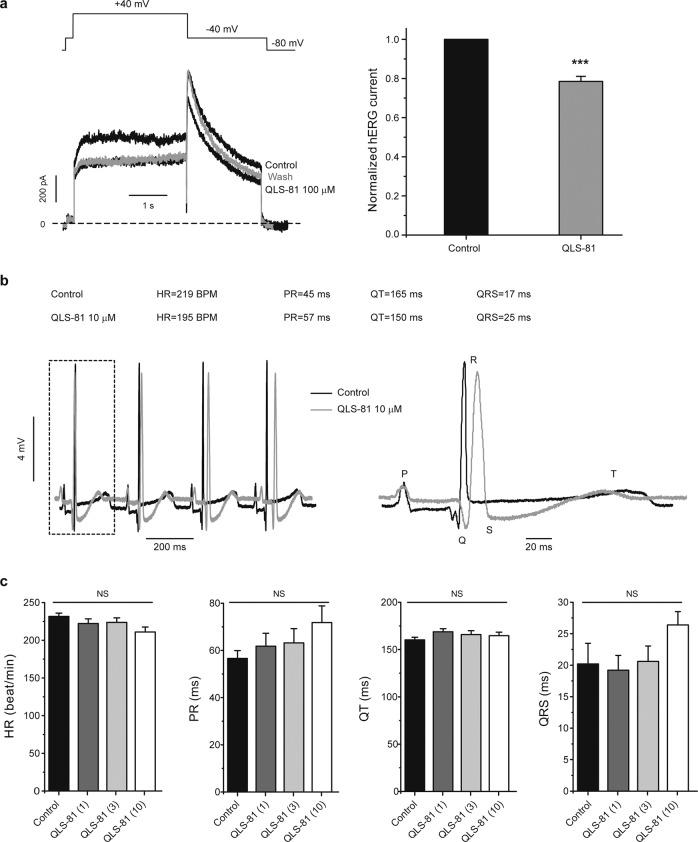
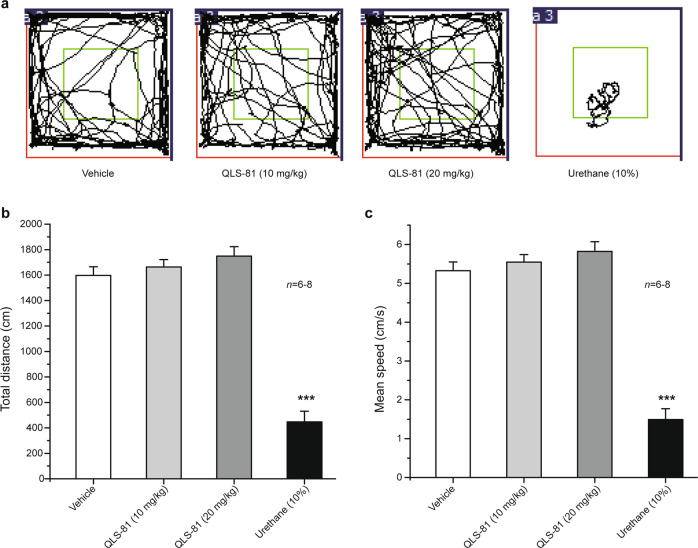
Voltage-gated sodium channel Nav1.7 robustly expressed in peripheral nociceptive neurons has been considered as a therapeutic target for chronic pain, but there is no selective Nav1.7 inhibitor available for therapy of chronic pain. Ralfinamide has shown anti-nociceptive activity in animal models of inflammatory and neuropathic pain and is currently under phase III clinical trial for neuropathic pain. Based on ralfinamide, a novel small molecule (S)-2-((3-(4-((2-fluorobenzyl) oxy) phenyl) propyl) amino) propanamide (QLS-81) was synthesized. Here, we report the electrophysiological and pharmacodynamic characterization of QLS-81 as a Nav1.7 channel inhibitor with promising anti-nociceptive activity. In whole-cell recordings of HEK293 cells stably expressing Nav1.7, QLS-81 (IC50 at 3.5 ± 1.5 μM) was ten-fold more potent than its parent compound ralfinamide (37.1 ± 2.9 μM) in inhibiting Nav1.7 current. QLS-81 inhibition on Nav1.7 current was use-dependent. Application of QLS-81 (10 μM) caused a hyperpolarizing shift of the fast and slow inactivation of Nav1.7 channel about 7.9 mV and 26.6 mV, respectively, and also slowed down the channel fast and slow inactivation recovery. In dissociated mouse DRG neurons, QLS-81 (10 μM) inhibited native Nav current and suppressed depolarizing current pulse-elicited neuronal firing. Administration of QLS-81 (2, 5, 10 mg· kg-1· d-1, i.p.) in mice for 10 days dose-dependently alleviated spinal nerve injury-induced neuropathic pain and formalin-induced inflammatory pain. In addition, QLS-81 (10 μM) did not significantly affect ECG in guinea pig heart ex vivo; and administration of QLS-81 (10, 20 mg/kg, i.p.) in mice had no significant effect on spontaneous locomotor activity. Taken together, our results demonstrate that QLS-81, as a novel Nav1.7 inhibitor, is efficacious on chronic pain in mice, and it may hold developmental potential for pain therapy.
Keywords: Nav1.7; QLS-81; anti-nociception; inflammatory pain; neuropathic pain; ralfinamide.
© 2021. The Author(s), under exclusive licence to CPS and SIMM.
Conflict of interest statement
The authors declare no competing interests.
Figures
Similar articles
-
Inhibition of TTX-S Na+ currents by a novel blocker QLS-278 for antinociception.J Pharmacol Exp Ther. 2025 Feb;392(2):100030. doi: 10.1124/jpet.124.002273. Epub 2024 Nov 22. J Pharmacol Exp Ther. 2025. PMID: 40023600
-
A novel isoquinoline alkaloid HJ-69 isolated from Zanthoxylum bungeanum attenuates inflammatory pain by inhibiting voltage-gated sodium and potassium channels.J Ethnopharmacol. 2024 Aug 10;330:118218. doi: 10.1016/j.jep.2024.118218. Epub 2024 Apr 25. J Ethnopharmacol. 2024. PMID: 38677570
-
DRG Voltage-Gated Sodium Channel 1.7 Is Upregulated in Paclitaxel-Induced Neuropathy in Rats and in Humans with Neuropathic Pain.J Neurosci. 2018 Jan 31;38(5):1124-1136. doi: 10.1523/JNEUROSCI.0899-17.2017. Epub 2017 Dec 18. J Neurosci. 2018. PMID: 29255002 Free PMC article.
-
Pain behavior in SCN9A (Nav1.7) and SCN10A (Nav1.8) mutant rodent models.Neurosci Lett. 2021 May 14;753:135844. doi: 10.1016/j.neulet.2021.135844. Epub 2021 Mar 26. Neurosci Lett. 2021. PMID: 33775738 Review.
-
Recent advances in small molecule Nav 1.7 inhibitors for cancer pain management.Bioorg Chem. 2024 Sep;150:107605. doi: 10.1016/j.bioorg.2024.107605. Epub 2024 Jun 29. Bioorg Chem. 2024. PMID: 38971095 Review.
Cited by
-
Long noncoding RNA small nucleolar RNA host gene 5 facilitates neuropathic pain in spinal nerve injury by promoting SCN9A expression via CDK9.Hum Cell. 2024 Mar;37(2):451-464. doi: 10.1007/s13577-023-01019-w. Epub 2024 Jan 2. Hum Cell. 2024. PMID: 38167752
-
Discordance between preclinical and clinical testing of Na V 1.7-selective inhibitors for pain.Pain. 2025 Mar 1;166(3):481-501. doi: 10.1097/j.pain.0000000000003425. Epub 2024 Oct 23. Pain. 2025. PMID: 39928833 Free PMC article. Review.
-
Vicious LQT induced by a combination of factors different from hERG inhibition.Front Pharmacol. 2022 Jul 22;13:930831. doi: 10.3389/fphar.2022.930831. eCollection 2022. Front Pharmacol. 2022. PMID: 35935820 Free PMC article.
-
Pathophysiology of Nociception and Rare Genetic Disorders with Increased Pain Threshold or Pain Insensitivity.Pathophysiology. 2022 Aug 2;29(3):435-452. doi: 10.3390/pathophysiology29030035. Pathophysiology. 2022. PMID: 35997391 Free PMC article. Review.
-
Suberoylanilide Hydroxamic Acid Ameliorates Pain Sensitization in Central Neuropathic Pain After Spinal Cord Injury via the HDAC5/NEDD4/SCN9A Axis.Neurochem Res. 2023 Aug;48(8):2436-2450. doi: 10.1007/s11064-023-03913-z. Epub 2023 Mar 31. Neurochem Res. 2023. PMID: 37002470
References
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
