

Admixed Populations Improve Power for Variant Discovery and Portability in Genome-Wide Association Studies
- PMID: 34108994
- PMCID: PMC8181458
- DOI: 10.3389/fgene.2021.673167
Admixed Populations Improve Power for Variant Discovery and Portability in Genome-Wide Association Studies
Abstract
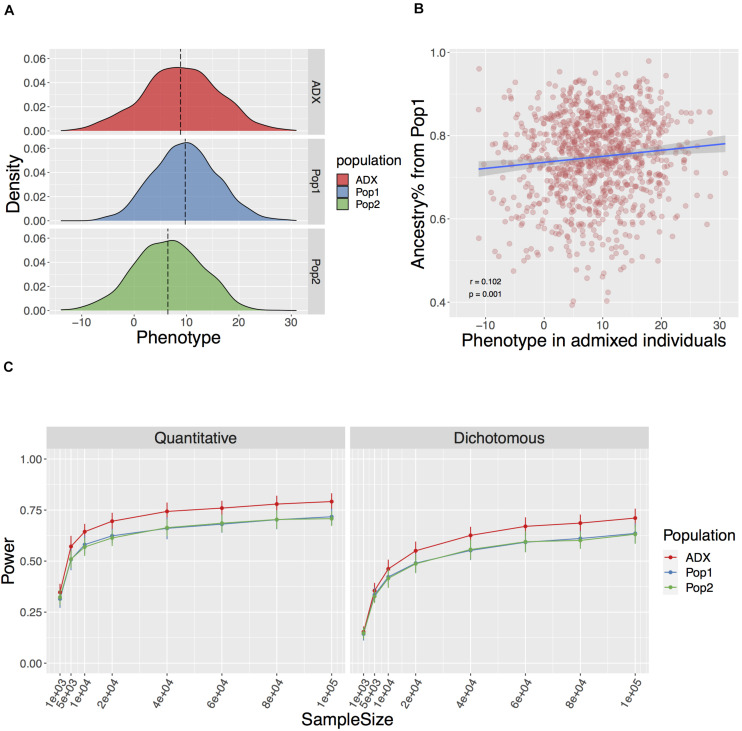
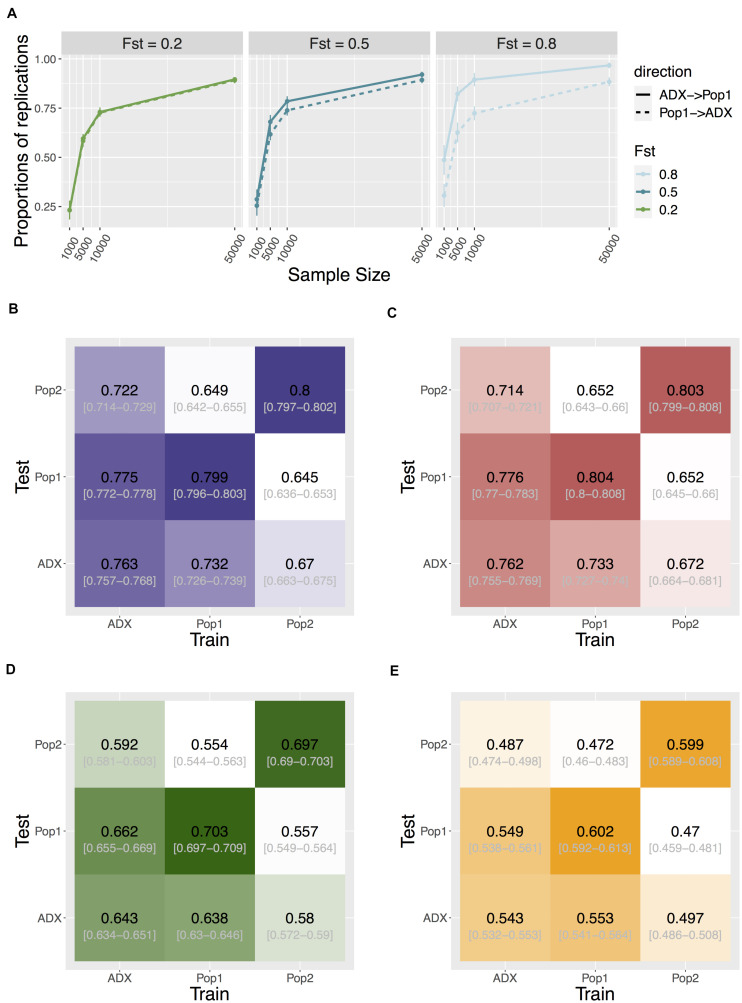
Genome-wide association studies (GWAS) are primarily conducted in single-ancestry settings. The low transferability of results has limited our understanding of human genetic architecture across a range of complex traits. In contrast to homogeneous populations, admixed populations provide an opportunity to capture genetic architecture contributed from multiple source populations and thus improve statistical power. Here, we provide a mechanistic simulation framework to investigate the statistical power and transferability of GWAS under directional polygenic selection or varying divergence. We focus on a two-way admixed population and show that GWAS in admixed populations can be enriched for power in discovery by up to 2-fold compared to the ancestral populations under similar sample size. Moreover, higher accuracy of cross-population polygenic score estimates is also observed if variants and weights are trained in the admixed group rather than in the ancestral groups. Common variant associations are also more likely to replicate if first discovered in the admixed group and then transferred to an ancestral population, than the other way around (across 50 iterations with 1,000 causal SNPs, training on 10,000 individuals, testing on 1,000 in each population, p = 3.78e-6, 6.19e-101, ∼0 for FST = 0.2, 0.5, 0.8, respectively). While some of these FST values may appear extreme, we demonstrate that they are found across the entire phenome in the GWAS catalog. This framework demonstrates that investigation of admixed populations harbors significant advantages over GWAS in single-ancestry cohorts for uncovering the genetic architecture of traits and will improve downstream applications such as personalized medicine across diverse populations.
Keywords: admixture; complex trait genetics; genetic architecture; polygenic score; statistical power.
Copyright © 2021 Lin, Park, Zaitlen, Henn and Gignoux.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures


References
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous