

Cholesterol Metabolic Reprogramming in Cancer and Its Pharmacological Modulation as Therapeutic Strategy
- PMID: 34109128
- PMCID: PMC8181394
- DOI: 10.3389/fonc.2021.682911
Cholesterol Metabolic Reprogramming in Cancer and Its Pharmacological Modulation as Therapeutic Strategy
Abstract
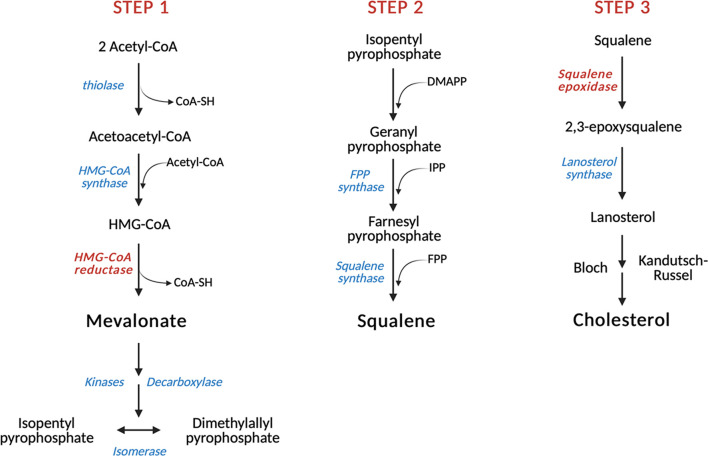
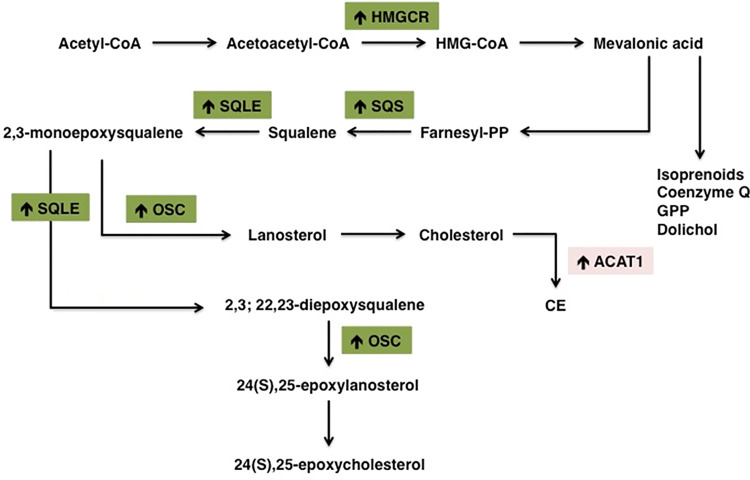
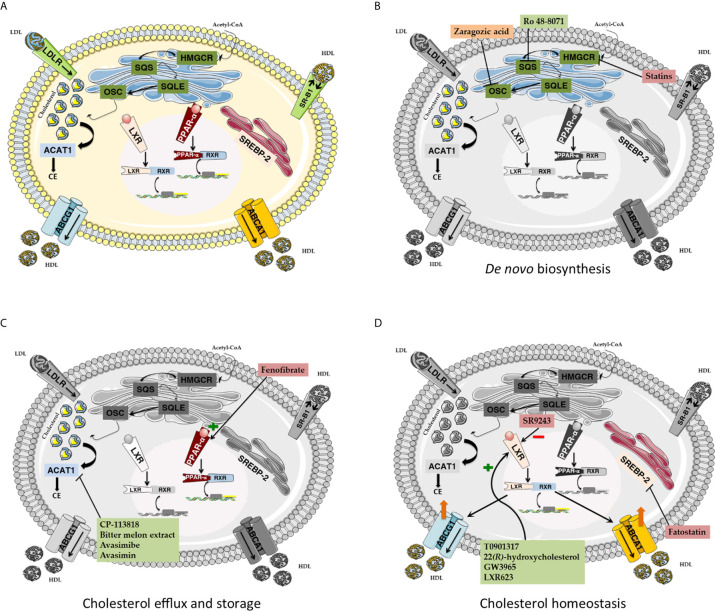
Cholesterol is a ubiquitous sterol with many biological functions, which are crucial for proper cellular signaling and physiology. Indeed, cholesterol is essential in maintaining membrane physical properties, while its metabolism is involved in bile acid production and steroid hormone biosynthesis. Additionally, isoprenoids metabolites of the mevalonate pathway support protein-prenylation and dolichol, ubiquinone and the heme a biosynthesis. Cancer cells rely on cholesterol to satisfy their increased nutrient demands and to support their uncontrolled growth, thus promoting tumor development and progression. Indeed, transformed cells reprogram cholesterol metabolism either by increasing its uptake and de novo biosynthesis, or deregulating the efflux. Alternatively, tumor can efficiently accumulate cholesterol into lipid droplets and deeply modify the activity of key cholesterol homeostasis regulators. In light of these considerations, altered pathways of cholesterol metabolism might represent intriguing pharmacological targets for the development of exploitable strategies in the context of cancer therapy. Thus, this work aims to discuss the emerging evidence of in vitro and in vivo studies, as well as clinical trials, on the role of cholesterol pathways in the treatment of cancer, starting from already available cholesterol-lowering drugs (statins or fibrates), and moving towards novel potential pharmacological inhibitors or selective target modulators.
Keywords: cancer; cancer therapy; cholesterol; metabolic reprogramming; metabolic targeting agents; pharmacological modulation; pharmacological targeting.
Copyright © 2021 Giacomini, Gianfanti, Desbats, Orso, Berretta, Prayer-Galetti, Ragazzi and Cocetta.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest. The reviewer AC declared a shared affiliation with the authors to the handling editor at the time of review.
Figures
References
Publication types
LinkOut - more resources
Full Text Sources