

Inflammatory signaling mechanisms in bipolar disorder
- PMID: 34112182
- PMCID: PMC8194019
- DOI: 10.1186/s12929-021-00742-6
Inflammatory signaling mechanisms in bipolar disorder
Abstract
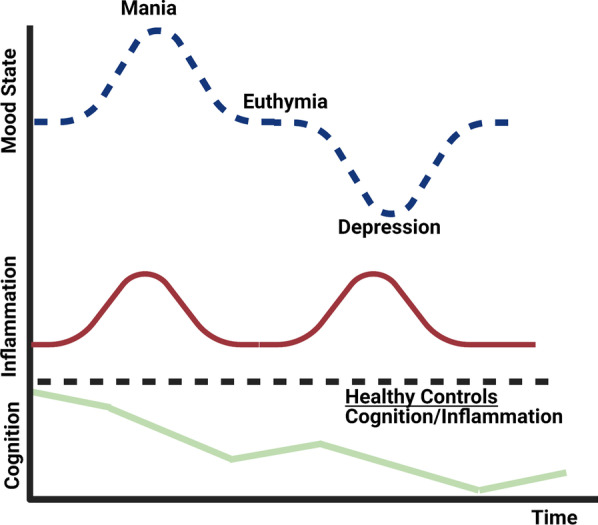
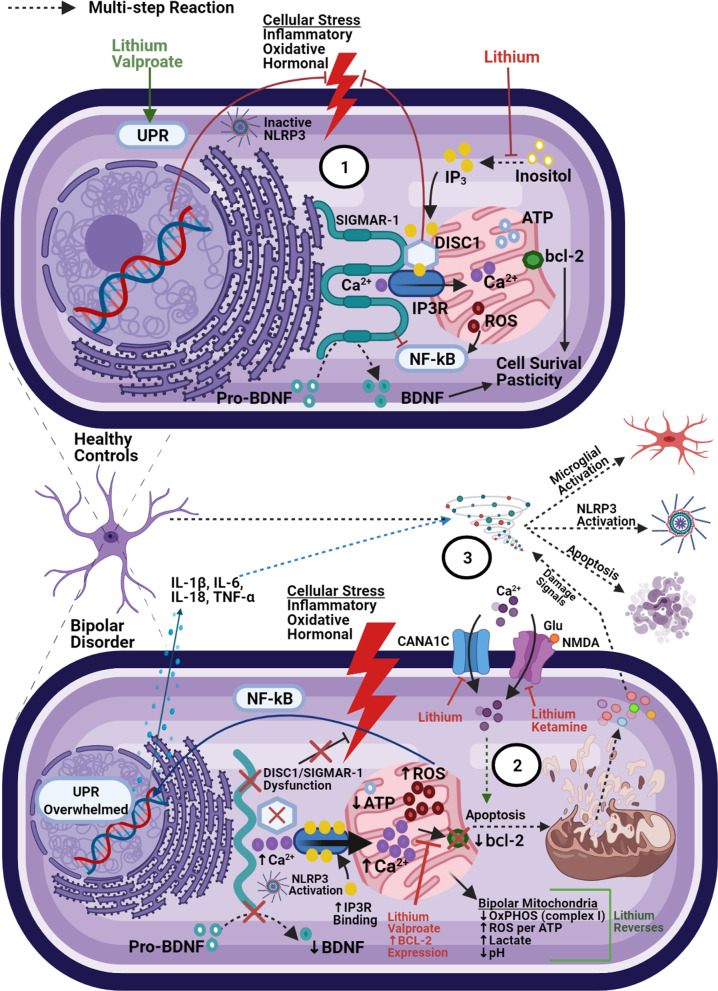
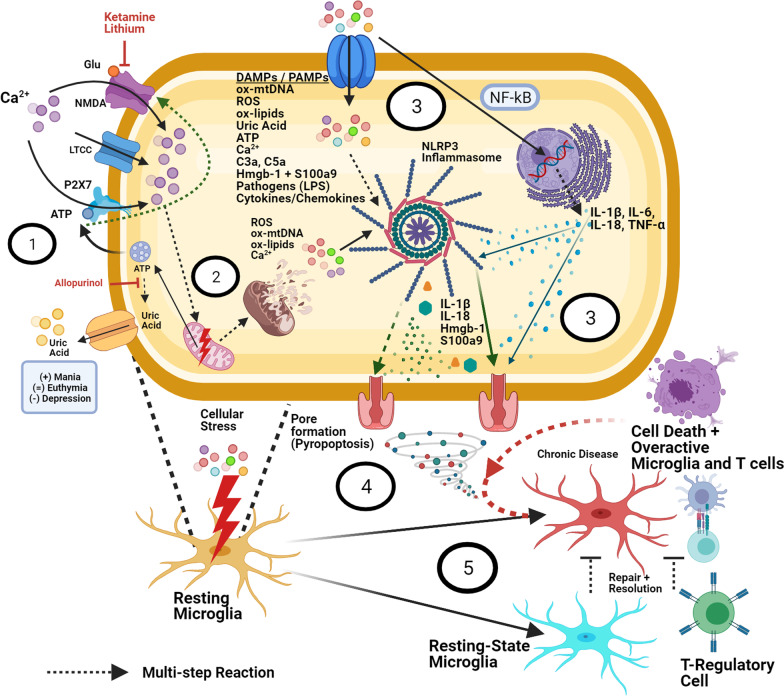
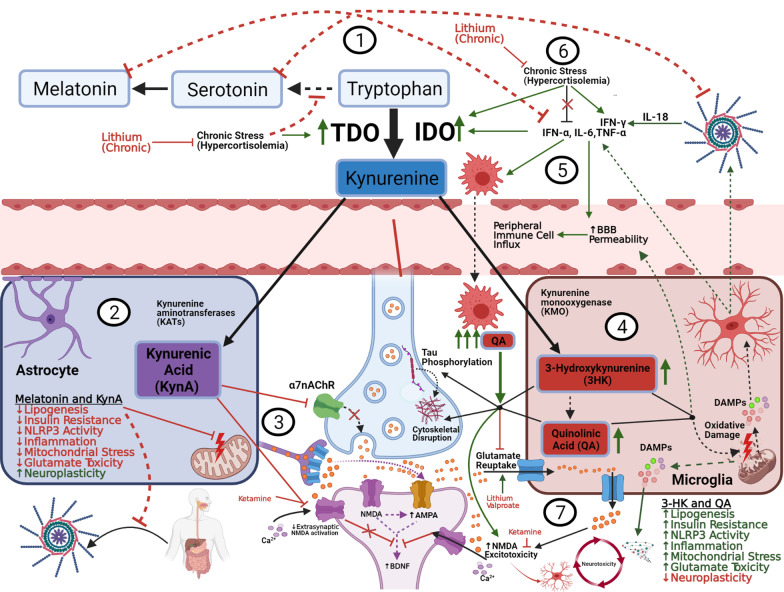
Bipolar disorder is a decidedly heterogeneous and multifactorial disease, with a high individual and societal burden. While not all patients display overt markers of elevated inflammation, significant evidence suggests that aberrant immune signaling contributes to all stages of the disease, and likely explains the elevated rates of comorbid inflammatory illnesses seen in this population. While individual systems have been intensely studied and targeted, a relative paucity of attention has been given to the interconnecting role of inflammatory signals therein. This review presents an updated overview of some of the most prominent pathophysiologic mechanisms in bipolar disorder, from mitochondrial, endoplasmic reticular, and calcium homeostasis, to purinergic, kynurenic, and hormonal/neurotransmitter signaling, showing inflammation to act as a powerful nexus between these systems. Several areas with a high degree of mechanistic convergence within this paradigm are highlighted to present promising future targets for therapeutic development and screening.
Keywords: BDNF; Bipolar; Glutamate; Inflammation; Mitochondria; NLRP3; P2X7; Purine.
Conflict of interest statement
The authors declare that they have no competing interests.
Figures
References
-
- Panaccione I, Spalletta G, Sani G. Neuroinflammation and excitatory symptoms in bipolar disorder. Neuroimmunol Neuroinflam. 2015;2(4):215–227. doi: 10.4103/2347-8659.167304. - DOI
