

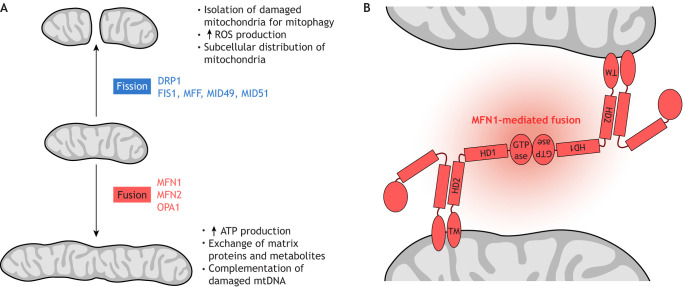
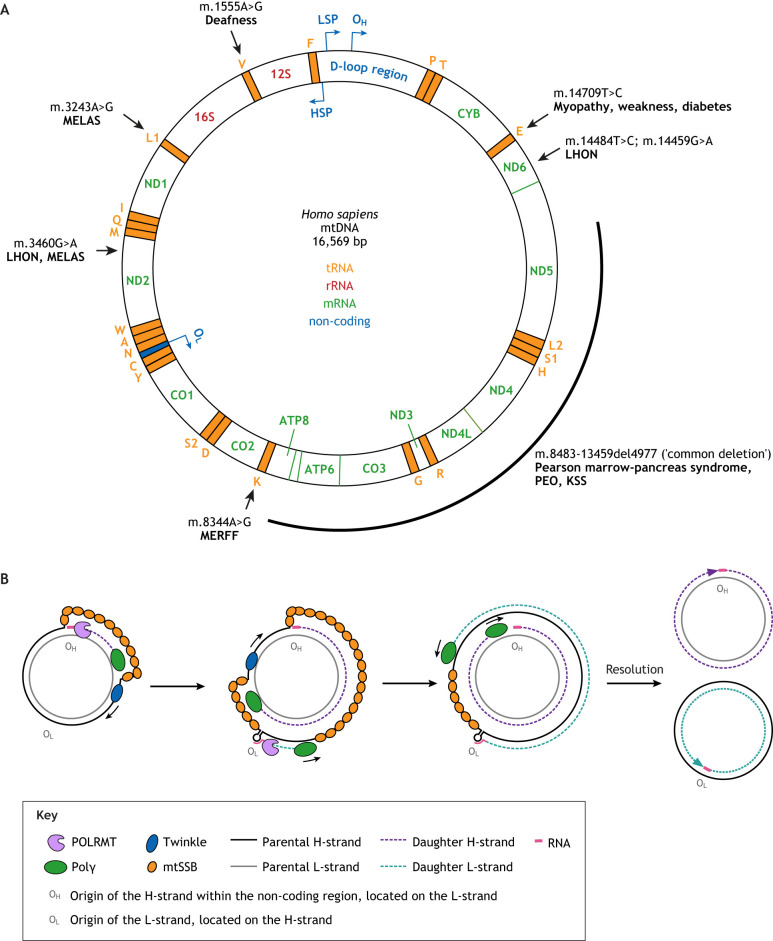
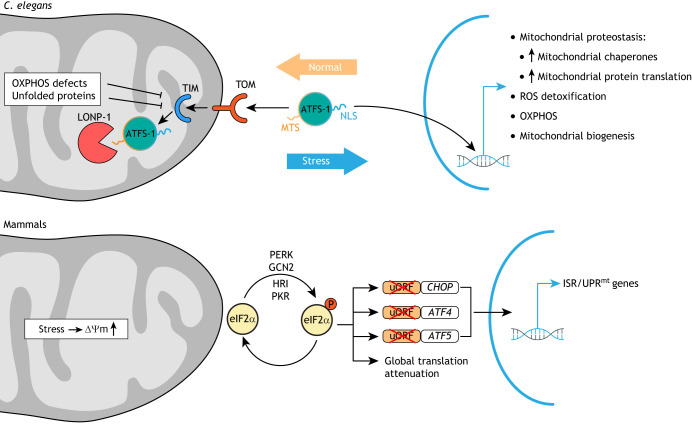
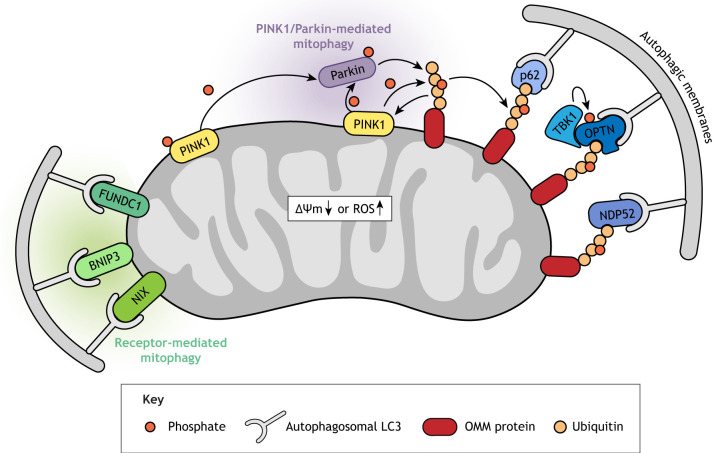
Mitochondrial function in development and disease
- PMID: 34114603
- PMCID: PMC8214736
- DOI: 10.1242/dmm.048912
Mitochondrial function in development and disease
Abstract
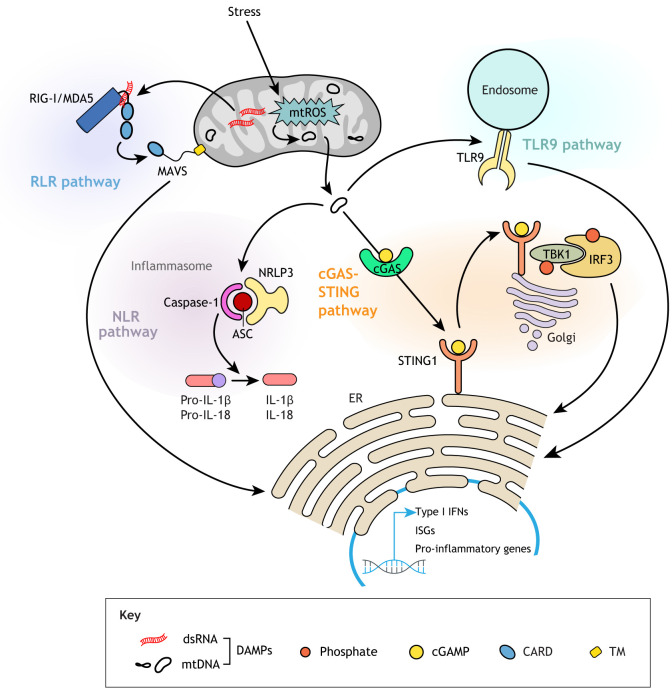
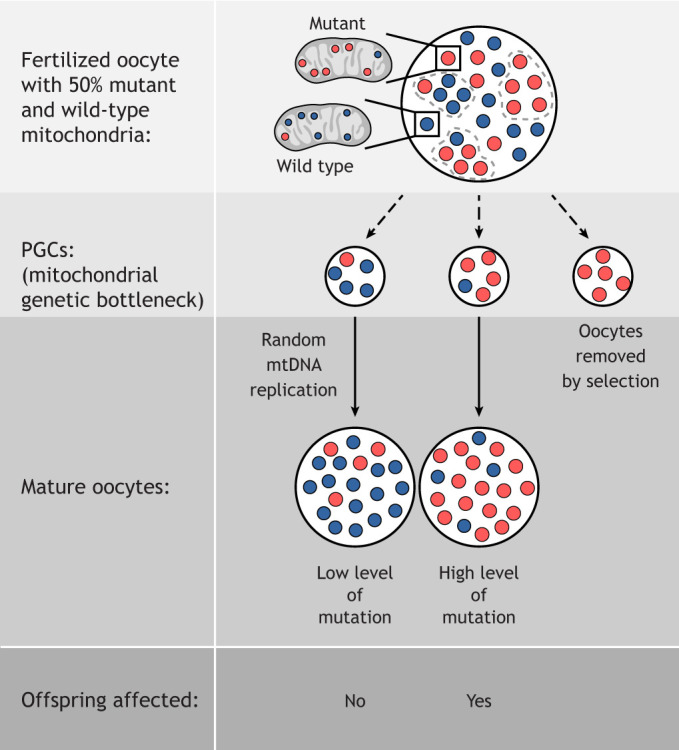
Mitochondria are organelles with vital functions in almost all eukaryotic cells. Often described as the cellular 'powerhouses' due to their essential role in aerobic oxidative phosphorylation, mitochondria perform many other essential functions beyond energy production. As signaling organelles, mitochondria communicate with the nucleus and other organelles to help maintain cellular homeostasis, allow cellular adaptation to diverse stresses, and help steer cell fate decisions during development. Mitochondria have taken center stage in the research of normal and pathological processes, including normal tissue homeostasis and metabolism, neurodegeneration, immunity and infectious diseases. The central role that mitochondria assume within cells is evidenced by the broad impact of mitochondrial diseases, caused by defects in either mitochondrial or nuclear genes encoding for mitochondrial proteins, on different organ systems. In this Review, we will provide the reader with a foundation of the mitochondrial 'hardware', the mitochondrion itself, with its specific dynamics, quality control mechanisms and cross-organelle communication, including its roles as a driver of an innate immune response, all with a focus on development, disease and aging. We will further discuss how mitochondrial DNA is inherited, how its mutation affects cell and organismal fitness, and current therapeutic approaches for mitochondrial diseases in both model organisms and humans.
Keywords: Mitochondrial diseases; Mitochondrial fusion and fission; Mitochondrial unfolded protein response; Mitophagy; mtDNA heteroplasmy and inheritance; mtDNA-mediated innate immune response.
© 2021. Published by The Company of Biologists Ltd.
Conflict of interest statement
Competing interests L.I.Z. is a founder of and holds stock in Fate Therapeutics, Camp4 Therapeutics and Scholar Rock, and is a consultant to Celularity. All other authors declare no competing interests.
Figures
References
-
- Abudayyeh, O. O., Gootenberg, J. S., Konermann, S., Joung, J., Slaymaker, I. M., Cox, D. B., Shmakov, S., Makarova, K. S., Semenova, E., Minakhin, L., et al. (2016). C2c2 is a single-component programmable RNA-guided RNA-targeting CRISPR effector. Science 353, aaf5573. 10.1126/science.aaf5573 - DOI - PMC - PubMed
-
- Acin-Perez, R., Bayona-Bafaluy, M. P., Bueno, M., Machicado, C., Fernandez-Silva, P., Perez-Martos, A., Montoya, J., Lopez-Perez, M. J., Sancho, J. and Enriquez, J. A. (2003). An intragenic suppressor in the cytochrome c oxidase I gene of mouse mitochondrial DNA. Hum. Mol. Genet. 12, 329-339. 10.1093/hmg/ddg021 - DOI - PubMed
-
- Aguirre, S., Luthra, P., Sanchez-Aparicio, M. T., Maestre, A. M., Patel, J., Lamothe, F., Fredericks, A. C., Tripathi, S., Zhu, T., Pintado-Silva, J., et al. (2017). Dengue virus NS2B protein targets cGAS for degradation and prevents mitochondrial DNA sensing during infection. Nat Microbiol 2, 17037. 10.1038/nmicrobiol.2017.37 - DOI - PMC - PubMed
-
- Ahlqvist, K. J., Hämäläinen, R. H., Yatsuga, S., Uutela, M., Terzioglu, M., Götz, A., Forsström, S., Salven, P., Angers-Loustau, A., Kopra, O. H., et al. (2012). Somatic progenitor cell vulnerability to mitochondrial DNA mutagenesis underlies progeroid phenotypes in Polg mutator mice. Cell Metab. 15, 100-109. 10.1016/j.cmet.2011.11.012 - DOI - PubMed
-
- Ahlqvist, K. J., Leoncini, S., Pecorelli, A., Wortmann, S. B., Ahola, S., Forsstrom, S., Guerranti, R., De Felice, C., Smeitink, J., Ciccoli, L., et al. (2015). MtDNA mutagenesis impairs elimination of mitochondria during erythroid maturation leading to enhanced erythrocyte destruction. Nat. Commun. 6, 6494. 10.1038/ncomms7494 - DOI - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
