

Open-Label Evaluation of Eteplirsen in Patients with Duchenne Muscular Dystrophy Amenable to Exon 51 Skipping: PROMOVI Trial
- PMID: 34120909
- PMCID: PMC8673535
- DOI: 10.3233/JND-210643
Open-Label Evaluation of Eteplirsen in Patients with Duchenne Muscular Dystrophy Amenable to Exon 51 Skipping: PROMOVI Trial
Abstract
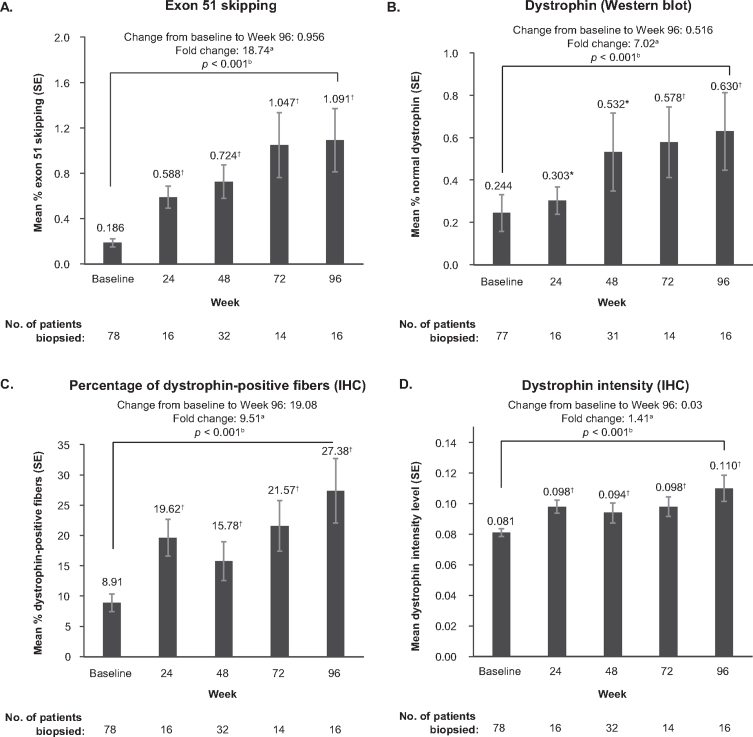
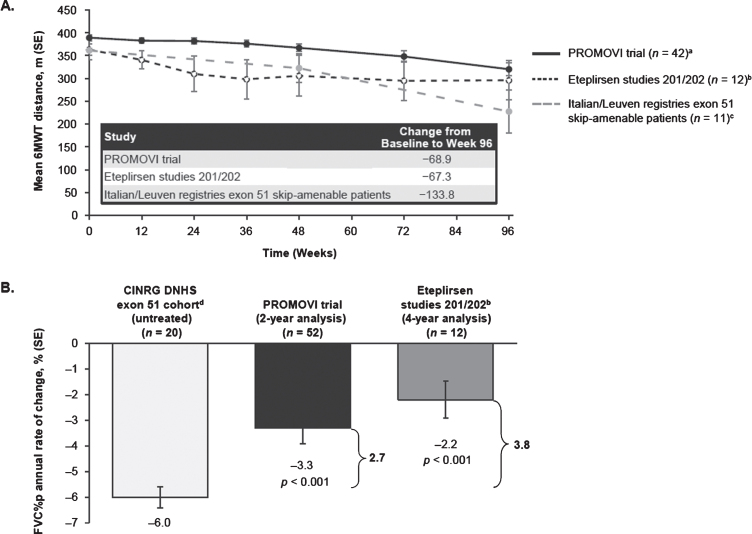
BackgroundEteplirsen received accelerated FDA approval for treatment of Duchenne muscular dystrophy (DMD) with mutations amenable to exon 51 skipping, based on demonstrated dystrophin production.ObjectiveTo report results from PROMOVI, a phase 3, multicenter, open-label study evaluating efficacy and safety of eteplirsen in a larger cohort.MethodsAmbulatory patients aged 7-16 years, with confirmed mutations amenable to exon 51 skipping, received eteplirsen 30 mg/kg/week intravenously for 96 weeks. An untreated cohort with DMD not amenable to exon 51 skipping was also enrolled.Results78/79 eteplirsen-treated patients completed 96 weeks of treatment. 15/30 untreated patients completed the study; this cohort was considered an inappropriate control group because of genotype-driven differences in clinical trajectory. At Week 96, eteplirsen-treated patients showed increased exon skipping (18.7-fold) and dystrophin protein (7-fold) versus baseline. Post-hoc comparisons with patients from eteplirsen phase 2 studies (4658-201/202) and mutation-matched external natural history controls confirmed previous results, suggesting clinically notable attenuation of decline on the 6-minute walk test over 96 weeks (PROMOVI: -68.9 m; phase 2 studies: -67.3 m; external controls: -133.8 m) and significant attenuation of percent predicted forced vital capacity annual decline (PROMOVI: -3.3%, phase 2 studies: -2.2%, external controls: -6.0%; p < 0.001). Adverse events were generally mild to moderate and unrelated to eteplirsen. Most frequent treatment-related adverse events were headache and vomiting; none led to treatment discontinuation.ConclusionsThis large, multicenter study contributes to the growing body of evidence for eteplirsen, confirming a positive treatment effect, favorable safety profile, and slowing of disease progression versus natural history.
Keywords: Muscular dystrophy; clinical trial; duchenne; phase 3; safety; treatment efficacy.
Conflict of interest statement
C. M. McDonald reports consulting (Astellas/Mitobridge, Bristol Myers Squibb, Capricor, Catabasis Pharmaceuticals, Edgewise Therapeutics, Eli Lilly, Epirium Bio [formerly Cardero Therapeutics], Gilead, Halo Therapeutics, Italfarmaco, Novartis, Pfizer, Prosensa, PTC Pharmaceuticals, Santhera Pharmaceuticals, and Sarepta Therapeutics, Inc.), and research funding, principal investigator, and speaking fees (Sarepta Therapeutics, Inc.). P. B. Shieh reports consulting fees for ad hoc advisory boards (AveXis, Biogen, PTC Therapeutics, and Sarepta Therapeutics, Inc.) and speakers’ bureau participation (Alexion, Biogen, and Grifols). H. Z. Abdel-Hamid reports advisory board participation (Audentes, AveXis, Biogen, and Sarepta Therapeutics, Inc.). A. M. Connolly reports advisory board participation (Acceleron, AveXis, Genentech, NS Pharma, Sarepta Therapeutics, Inc. and Scholar Rock) and DMSB participation (Catabasis). E. Ciafaloni reports advisory board participation (AveXis, Biogen, and Sarepta Therapeutics, Inc.). K. R. Wagner reports consulting fees (AskBio, Dyne, Vita, Dynacure, PTC Therapeutics, Roche, and Sarepta Therapeutics, Inc). N. Goemans reports advisory board participations and/or speaking fees (Italfarmaco, Biogen, Roche, AveXis, Pfizer, PTC Pharmaceuticals, Santhera Pharmaceuticals, and Sarepta Therapeutics, Inc.). E. Mercuri reports paid consulting (Sarepta Therapeutics, Inc.). J. R. Mendell reports receiving grants (Parent Project Muscular Dystrophy) and consulting fees (Sarepta Therapeutics, Inc. and Nationwide Children’s Hospital). N. Khan, E. Koenig, J. Malhotra, W. Zhang, and B. Han are employees of Sarepta Therapeutics, Inc. and own stock/options in the company.
Figures
References
-
- Birnkrant DJ, Bushby K, Bann CM, Apkon SD, Blackwell A, Brumbaugh D, et al. Diagnosis and management of Duchenne muscular dystrophy, part diagnosis, and neuromuscular, rehabilitation, endocrine, and gastrointestinal and nutritional management. Lancet Neurol. 2018;17(3):251–67. doi: 10.1016/S1474-4422(18)30024-3 - DOI - PMC - PubMed
