

DP4-AI automated NMR data analysis: straight from spectrometer to structure
- PMID: 34122893
- PMCID: PMC8152620
- DOI: 10.1039/d0sc00442a
DP4-AI automated NMR data analysis: straight from spectrometer to structure
Abstract
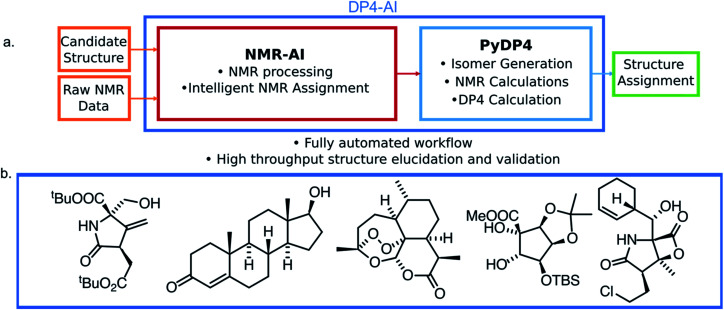
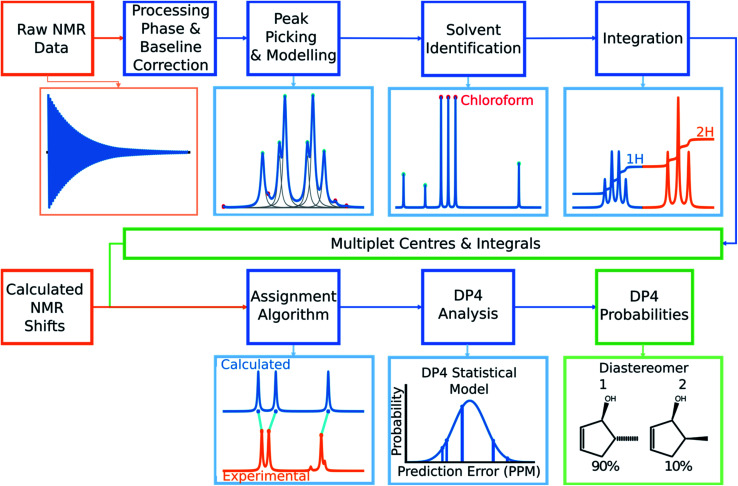
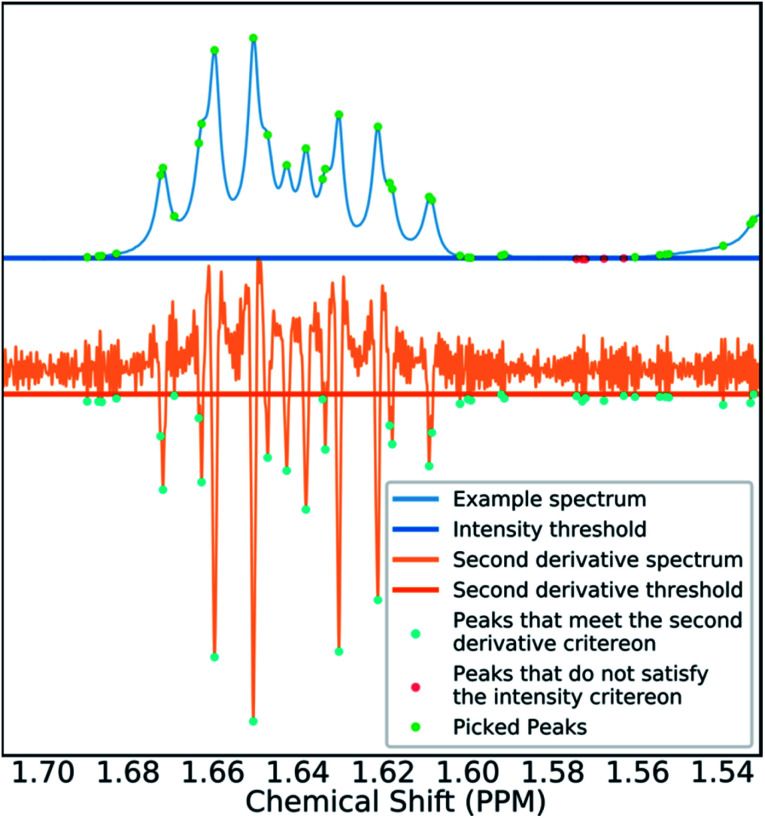
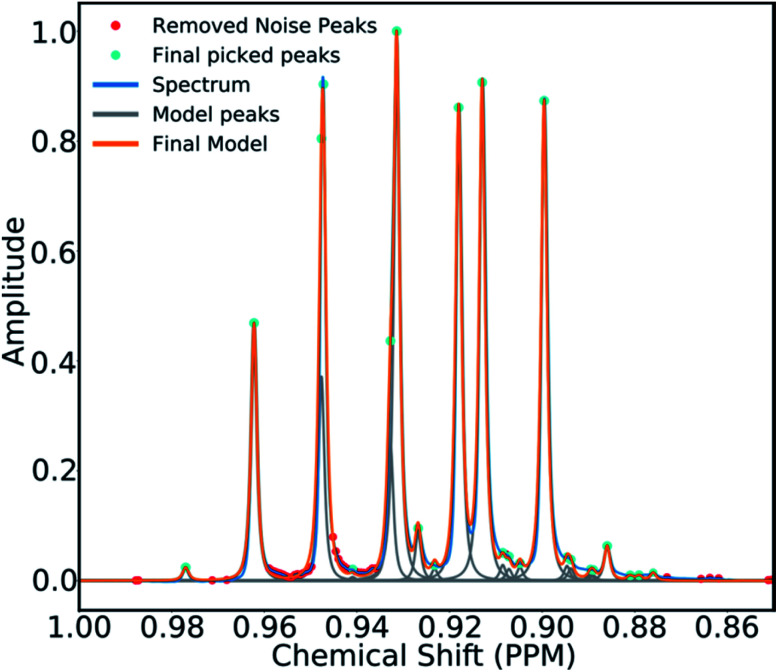
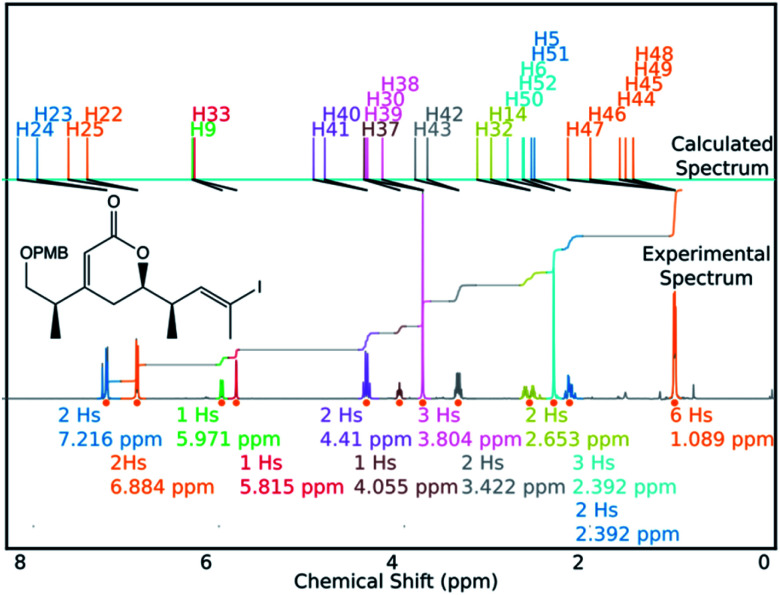
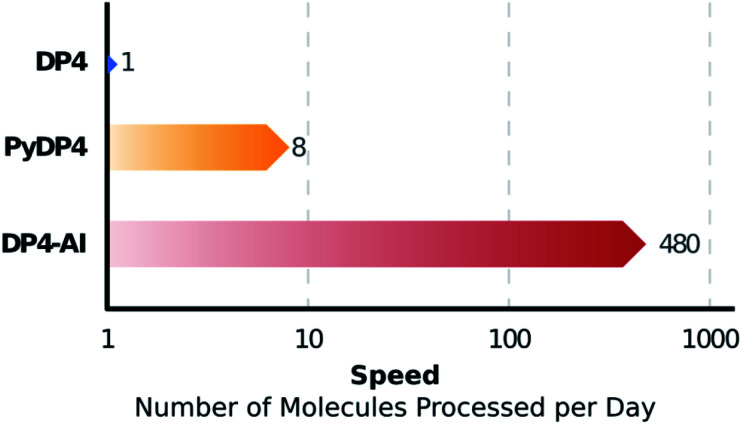
A robust system for automatic processing and assignment of raw 13C and 1H NMR data DP4-AI has been developed and integrated into our computational organic molecule structure elucidation workflow. Starting from a molecular structure with undefined stereochemistry or other structural uncertainty, this system allows for completely automated structure elucidation. Methods for NMR peak picking using objective model selection and algorithms for matching the calculated 13C and 1H NMR shifts to peaks in noisy experimental NMR data were developed. DP4-AI achieved a 60-fold increase in processing speed, and near-elimination of the need for scientist time, when rigorously evaluated using a challenging test set of molecules. DP4-AI represents a leap forward in NMR structure elucidation and a step-change in the functionality of DP4. It enables high-throughput analyses of databases and large sets of molecules, which were previously impossible, and paves the way for the discovery of new structural information through machine learning. This new functionality has been coupled with an intuitive GUI and is available as open-source software at https://github.com/KristapsE/DP4-AI.
This journal is © The Royal Society of Chemistry.
Conflict of interest statement
There are no conflicts to declare.
Figures
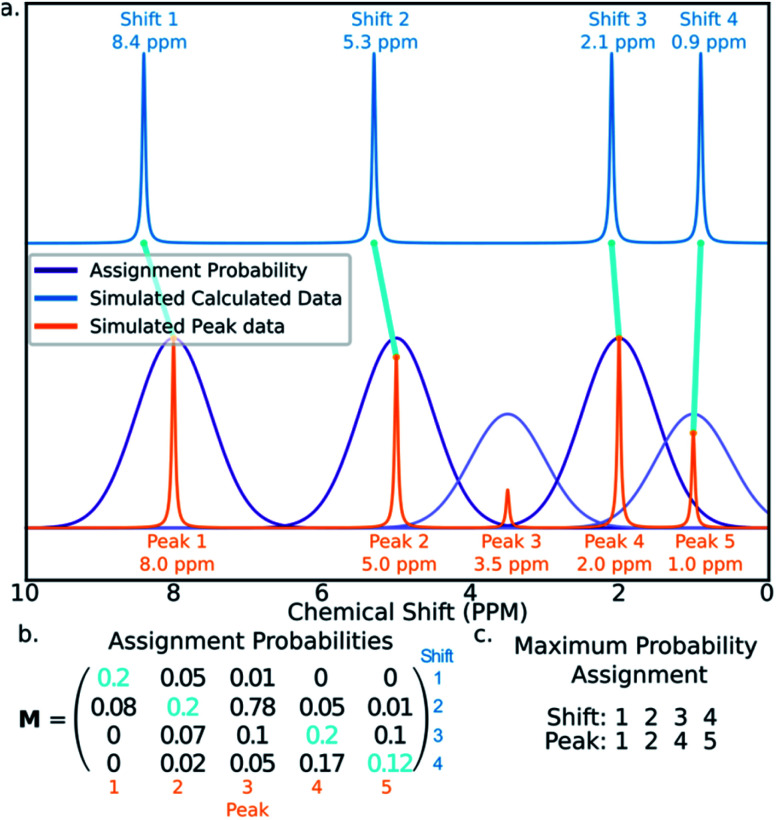
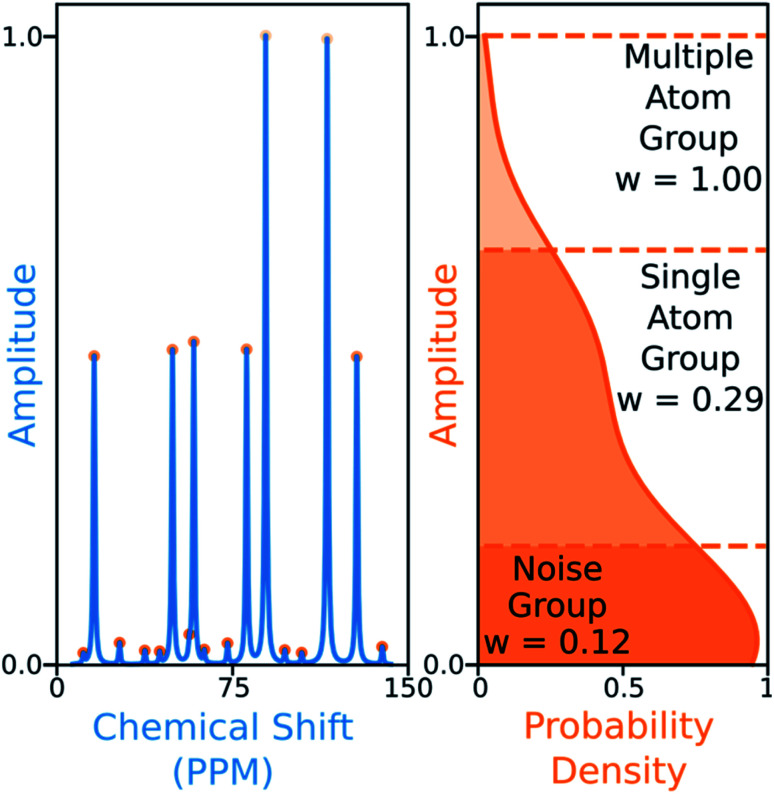
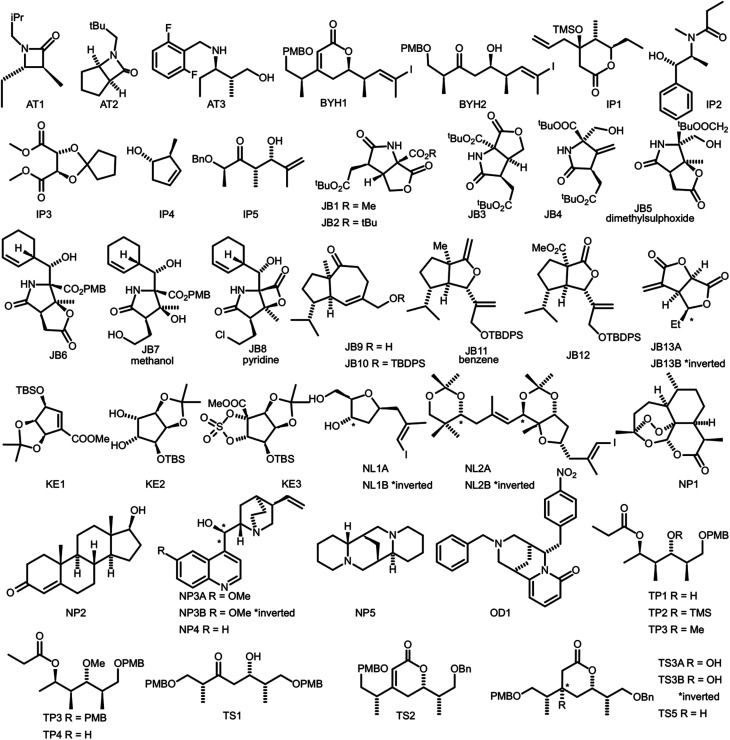
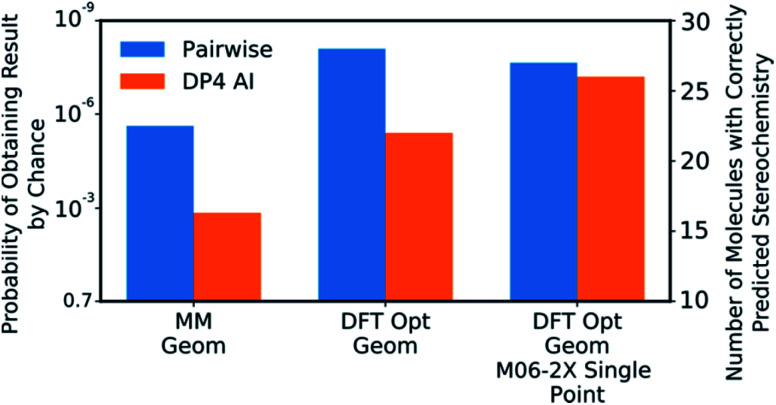
Similar articles
-
The DP5 probability, quantification and visualisation of structural uncertainty in single molecules.Chem Sci. 2022 Feb 25;13(12):3507-3518. doi: 10.1039/d1sc04406k. eCollection 2022 Mar 24. Chem Sci. 2022. PMID: 35432857 Free PMC article.
-
NMR Calculations with Quantum Methods: Development of New Tools for Structural Elucidation and Beyond.Acc Chem Res. 2020 Sep 15;53(9):1922-1932. doi: 10.1021/acs.accounts.0c00365. Epub 2020 Aug 14. Acc Chem Res. 2020. PMID: 32794691
-
When not to rely on Boltzmann populations. Automated CASE-3D structure elucidation of hyacinthacines through chemical shift differences.Magn Reson Chem. 2020 Feb;58(2):139-144. doi: 10.1002/mrc.4951. Epub 2019 Nov 10. Magn Reson Chem. 2020. PMID: 31663628
-
A critical review on the use of DP4+ in the structural elucidation of natural products: the good, the bad and the ugly. A practical guide.Nat Prod Rep. 2022 Jan 26;39(1):58-76. doi: 10.1039/d1np00030f. Nat Prod Rep. 2022. PMID: 34212963 Review.
-
Weizhouochrones: Gorgonian-Derived Symmetric Dimers and Their Structure Elucidation Using Anisotropic NMR Combined with DP4+ Probability and CASE-3D.J Nat Prod. 2022 Jul 22;85(7):1730-1737. doi: 10.1021/acs.jnatprod.2c00257. Epub 2022 Jul 6. J Nat Prod. 2022. PMID: 35792821 Review.
Cited by
-
Artificial intelligence in microbial natural product drug discovery: current and emerging role.Nat Prod Rep. 2022 Dec 14;39(12):2215-2230. doi: 10.1039/d2np00035k. Nat Prod Rep. 2022. PMID: 36017693 Free PMC article. Review.
-
Heterologous production of cyanobacterial compounds.J Ind Microbiol Biotechnol. 2021 Jun 4;48(3-4):kuab003. doi: 10.1093/jimb/kuab003. J Ind Microbiol Biotechnol. 2021. PMID: 33928376 Free PMC article. Review.
-
Structural Investigation of Aaptourinamine by a Novel Module-Assembly-Based Calculation.Mar Drugs. 2022 Oct 20;20(10):649. doi: 10.3390/md20100649. Mar Drugs. 2022. PMID: 36286473 Free PMC article.
-
Interpreting vibrational circular dichroism spectra: the Cai•factor for absolute configuration with confidence.J Cheminform. 2023 Mar 21;15(1):36. doi: 10.1186/s13321-023-00706-y. J Cheminform. 2023. PMID: 36945031 Free PMC article.
-
Real-time prediction of 1H and 13C chemical shifts with DFT accuracy using a 3D graph neural network.Chem Sci. 2021 Aug 9;12(36):12012-12026. doi: 10.1039/d1sc03343c. eCollection 2021 Sep 22. Chem Sci. 2021. PMID: 34667567 Free PMC article.
References
LinkOut - more resources
Full Text Sources