

Exploring the Binding Interaction of Raf Kinase Inhibitory Protein With the N-Terminal of C-Raf Through Molecular Docking and Molecular Dynamics Simulation
- PMID: 34124147
- PMCID: PMC8194344
- DOI: 10.3389/fmolb.2021.655035
Exploring the Binding Interaction of Raf Kinase Inhibitory Protein With the N-Terminal of C-Raf Through Molecular Docking and Molecular Dynamics Simulation
Abstract
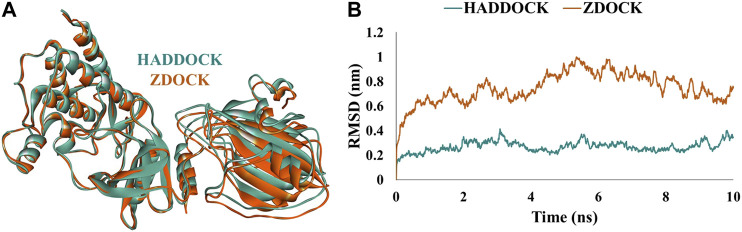
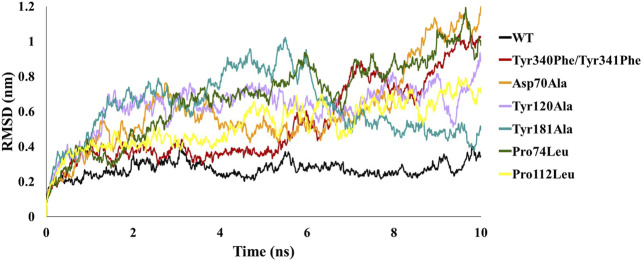
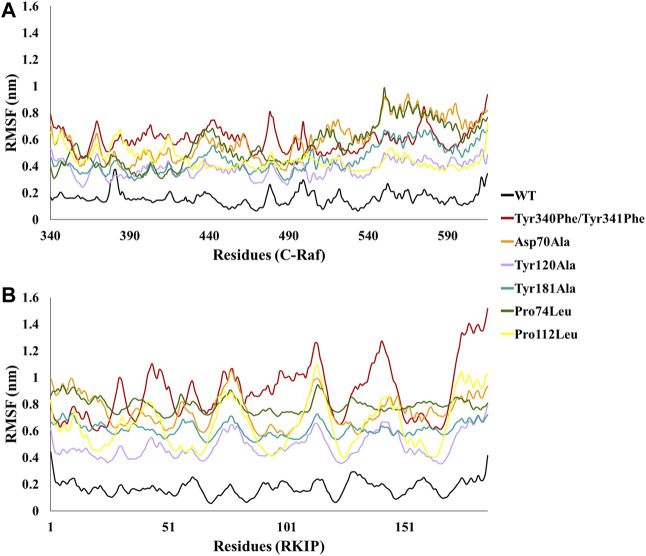
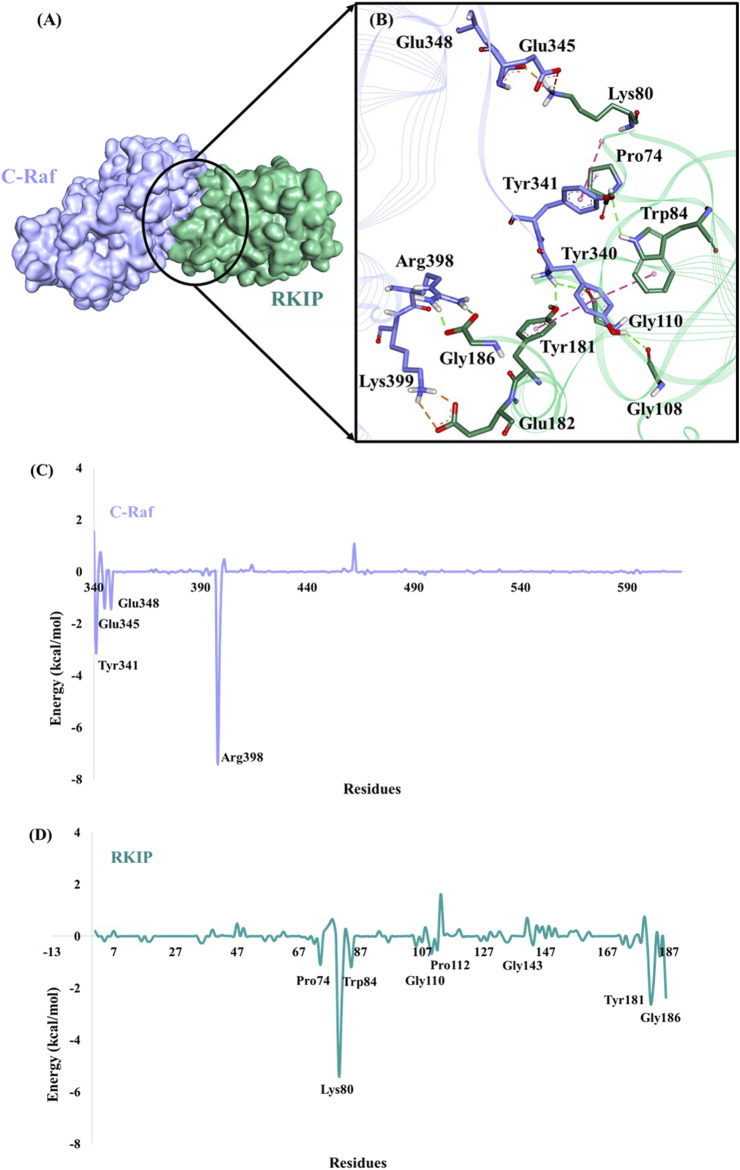
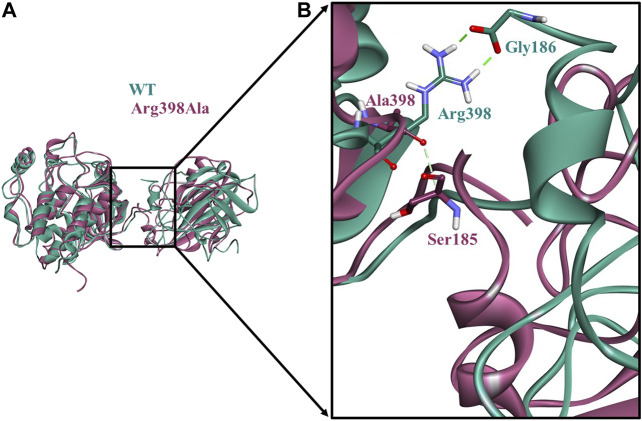
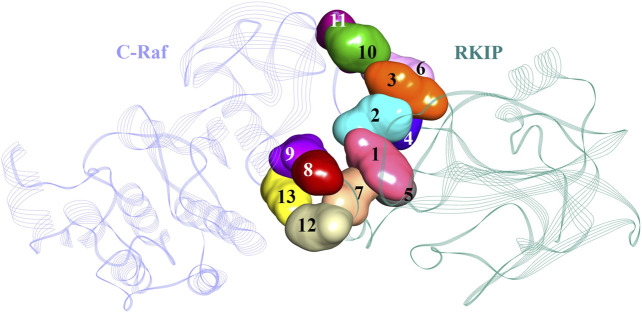
Protein-protein interactions are indispensable physiological processes regulating several biological functions. Despite the availability of structural information on protein-protein complexes, deciphering their complex topology remains an outstanding challenge. Raf kinase inhibitory protein (RKIP) has gained substantial attention as a favorable molecular target for numerous pathologies including cancer and Alzheimer's disease. RKIP interferes with the RAF/MEK/ERK signaling cascade by endogenously binding with C-Raf (Raf-1 kinase) and preventing its activation. In the current investigation, the binding of RKIP with C-Raf was explored by knowledge-based protein-protein docking web-servers including HADDOCK and ZDOCK and a consensus binding mode of C-Raf/RKIP structural complex was obtained. Molecular dynamics (MD) simulations were further performed in an explicit solvent to sample the conformations for when RKIP binds to C-Raf. Some of the conserved interface residues were mutated to alanine, phenylalanine and leucine and the impact of mutations was estimated by additional MD simulations and MM/PBSA analysis for the wild-type (WT) and constructed mutant complexes. Substantial decrease in binding free energy was observed for the mutant complexes as compared to the binding free energy of WT C-Raf/RKIP structural complex. Furthermore, a considerable increase in average backbone root mean square deviation and fluctuation was perceived for the mutant complexes. Moreover, per-residue energy contribution analysis of the equilibrated simulation trajectory by HawkDock and ANCHOR web-servers was conducted to characterize the key residues for the complex formation. One residue each from C-Raf (Arg398) and RKIP (Lys80) were identified as the druggable "hot spots" constituting the core of the binding interface and corroborated by additional long-time scale (300 ns) MD simulation of Arg398Ala mutant complex. A notable conformational change in Arg398Ala mutant occurred near the mutation site as compared to the equilibrated C-Raf/RKIP native state conformation and an essential hydrogen bonding interaction was lost. The thirteen binding sites assimilated from the overall analysis were mapped onto the complex as surface and divided into active and allosteric binding sites, depending on their location at the interface. The acquired information on the predicted 3D structural complex and the detected sites aid as promising targets in designing novel inhibitors to block the C-Raf/RKIP interaction.
Keywords: C-Raf; HADDOCK; MM/PBSA; RKIP; ZDOCK; binding sites prediction; molecular dynamics simulation; protein-protein docking.
Copyright © 2021 Parate, Rampogu, Lee, Hong and Lee.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures
References
-
- Abraham M. J., Murtola T., Schulz R., Páll S., Smith J. C., Hess B., et al. (2015). Gromacs: High Performance Molecular Simulations through Multi-Level Parallelism from Laptops to Supercomputers. SoftwareX. 1-2 (2), 19–25. 10.1016/j.softx.2015.06.001 - DOI
-
- Beshir A. B., Argueta C. E., Menikarachchi L. C., Gascon J. A., Fenteany G., Fenteany G. (2011). Locostatin Disrupts Association of Raf Kinase Inhibitor Protein with Binding Proteins by Modifying a Conserved Histidine Residue in the Ligand-Binding Pocket. Forum Immun. Dis. Ther. 2, 47–58. 10.1615/forumimmundisther.v2.i1.60 - DOI - PMC - PubMed
-
- Chen F., Liu H., Sun H., Pan P., Li Y., Li D., et al. (2016). Assessing the Performance of the MM/PBSA and MM/GBSA Methods. 6. Capability to Predict Protein-Protein Binding Free Energies and Re-rank Binding Poses Generated by Protein-Protein Docking. Phys. Chem. Chem. Phys. 18, 22129–22139. 10.1039/c6cp03670h - DOI - PubMed
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous