

PI3K inhibitors are finally coming of age
- PMID: 34127844
- PMCID: PMC9297732
- DOI: 10.1038/s41573-021-00209-1
PI3K inhibitors are finally coming of age
Erratum in
-
Author Correction: PI3K inhibitors are finally coming of age.Nat Rev Drug Discov. 2021 Oct;20(10):798. doi: 10.1038/s41573-021-00300-7. Nat Rev Drug Discov. 2021. PMID: 34471263 No abstract available.
Abstract
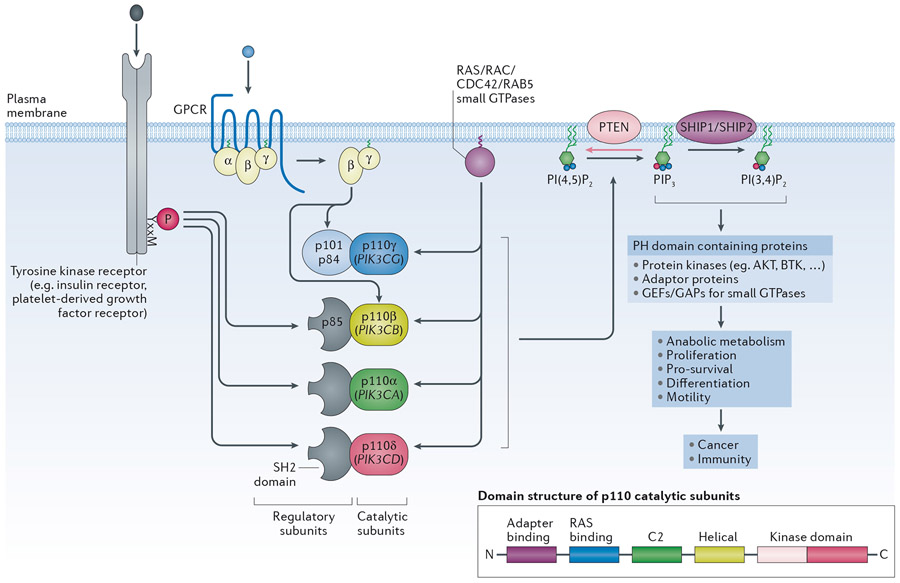
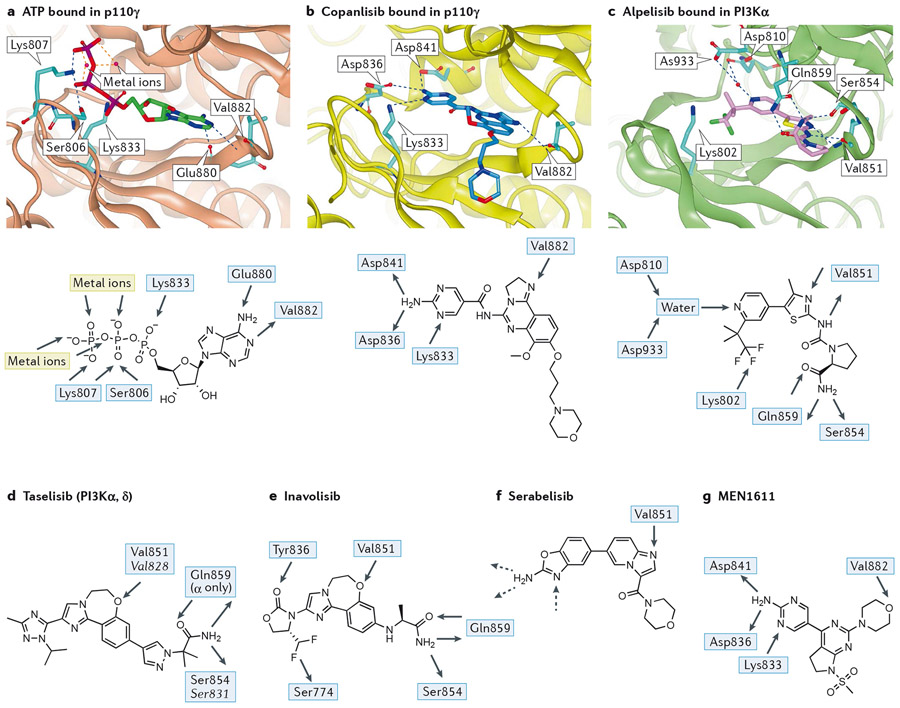
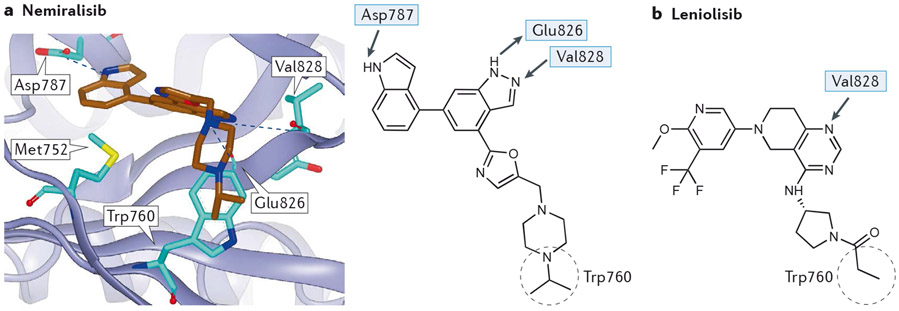
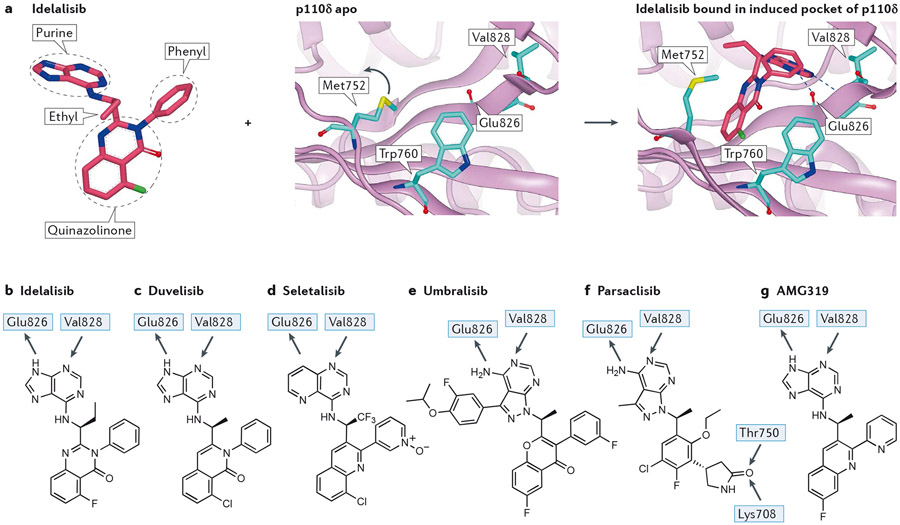
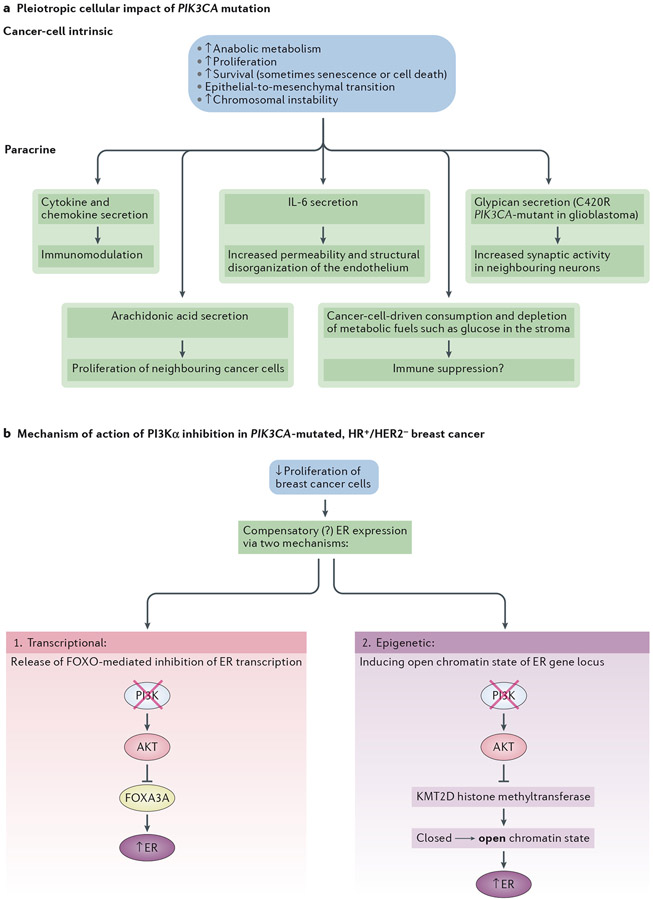
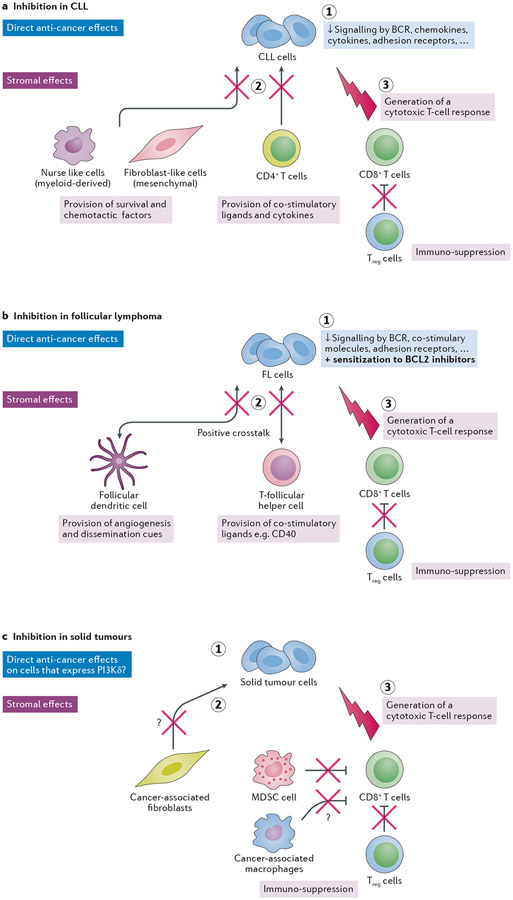
Overactive phosphoinositide 3-kinase (PI3K) in cancer and immune dysregulation has spurred extensive efforts to develop therapeutic PI3K inhibitors. Although progress has been hampered by issues such as poor drug tolerance and drug resistance, several PI3K inhibitors have now received regulatory approval - the PI3Kα isoform-selective inhibitor alpelisib for the treatment of breast cancer and inhibitors mainly aimed at the leukocyte-enriched PI3Kδ in B cell malignancies. In addition to targeting cancer cell-intrinsic PI3K activity, emerging evidence highlights the potential of PI3K inhibitors in cancer immunotherapy. This Review summarizes key discoveries that aid the clinical translation of PI3Kα and PI3Kδ inhibitors, highlighting lessons learnt and future opportunities.
© 2021. Springer Nature Limited.
Conflict of interest statement
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
