

Eruptive xanthoma model reveals endothelial cells internalize and metabolize chylomicrons, leading to extravascular triglyceride accumulation
- PMID: 34128469
- PMCID: PMC8203467
- DOI: 10.1172/JCI145800
Eruptive xanthoma model reveals endothelial cells internalize and metabolize chylomicrons, leading to extravascular triglyceride accumulation
Abstract
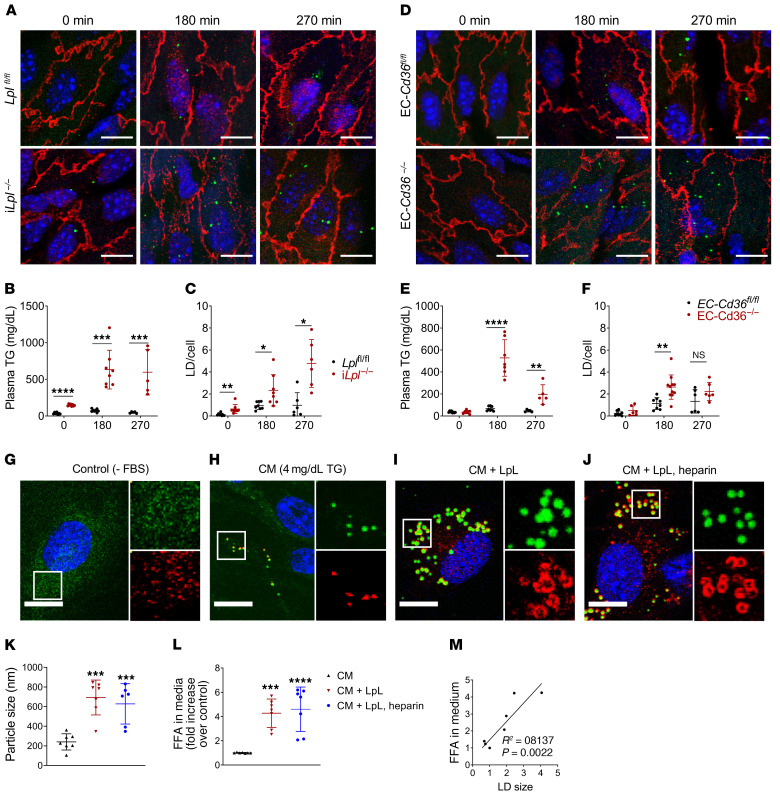
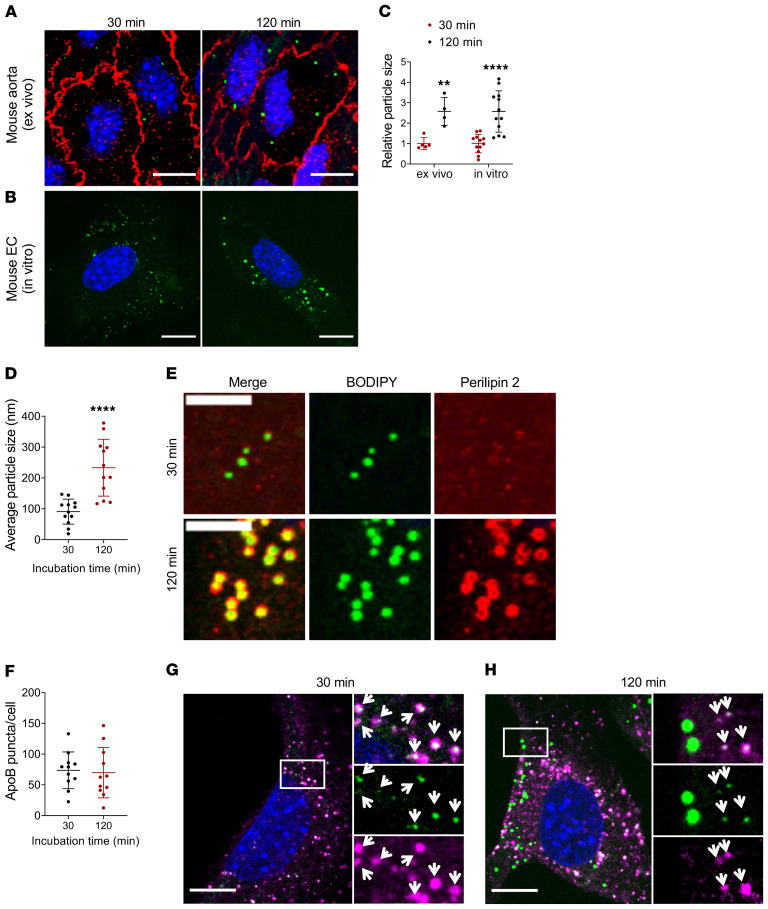
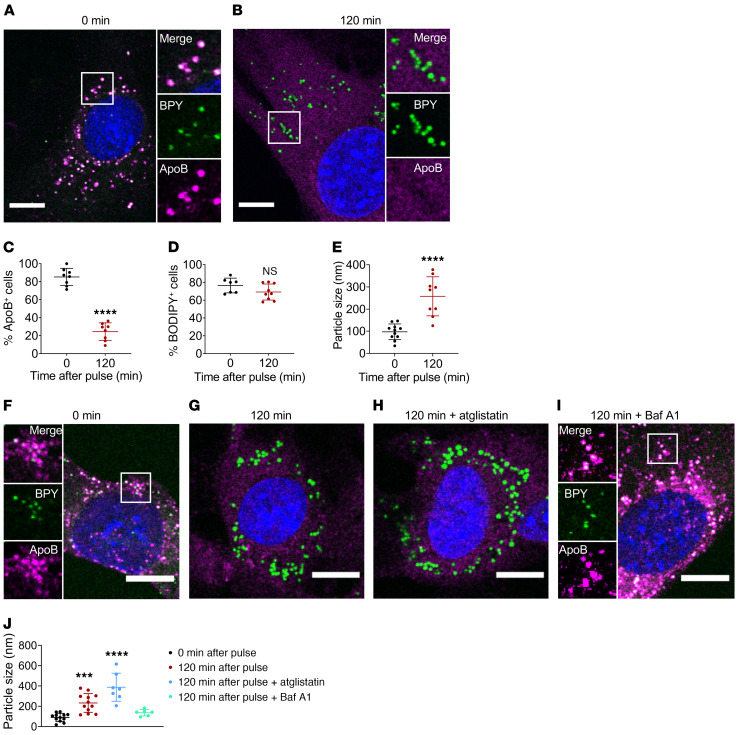
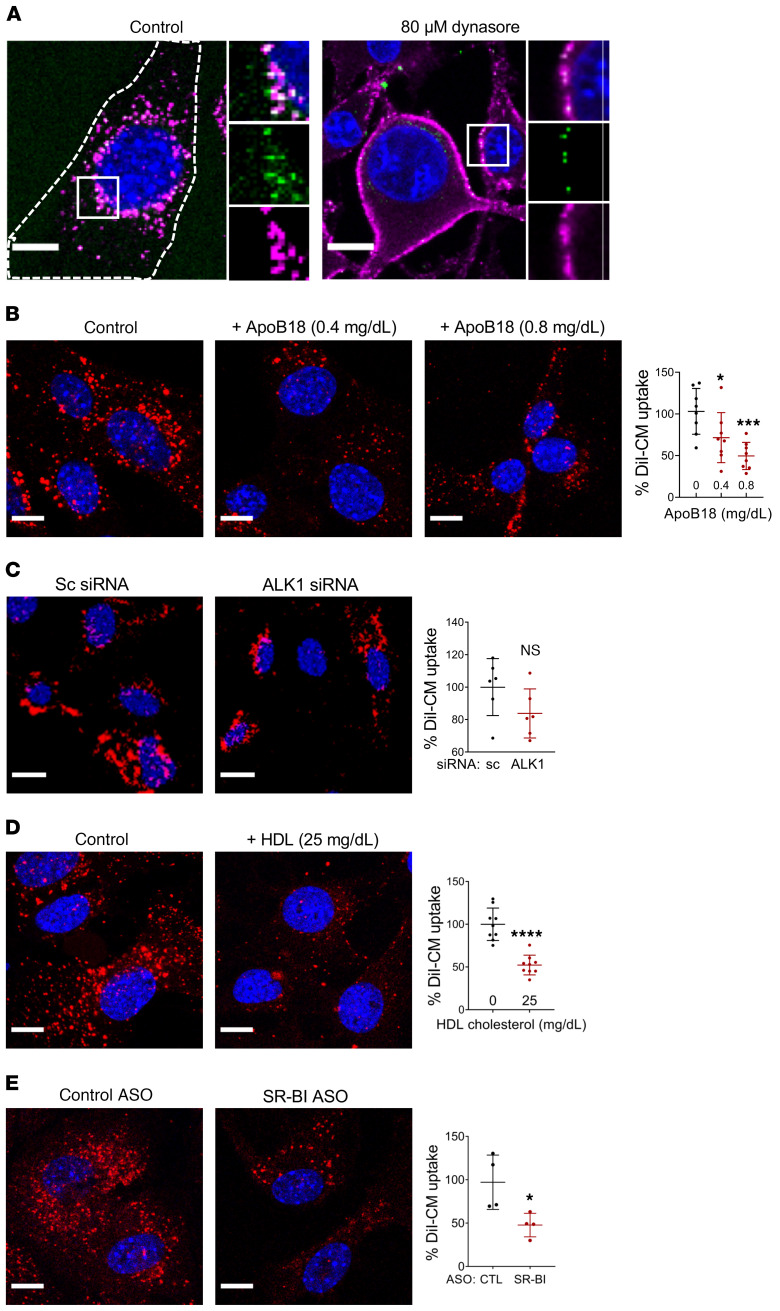
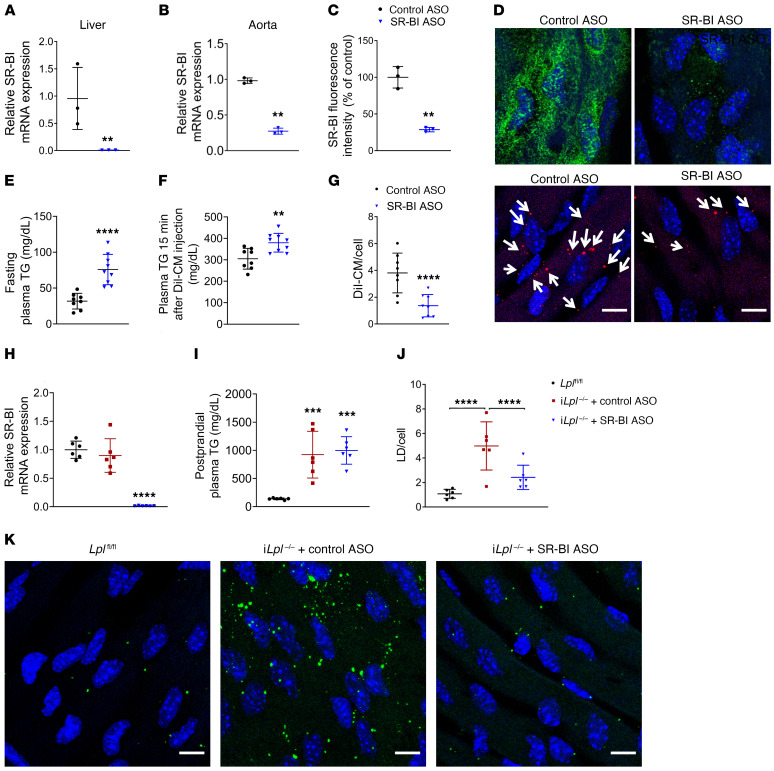
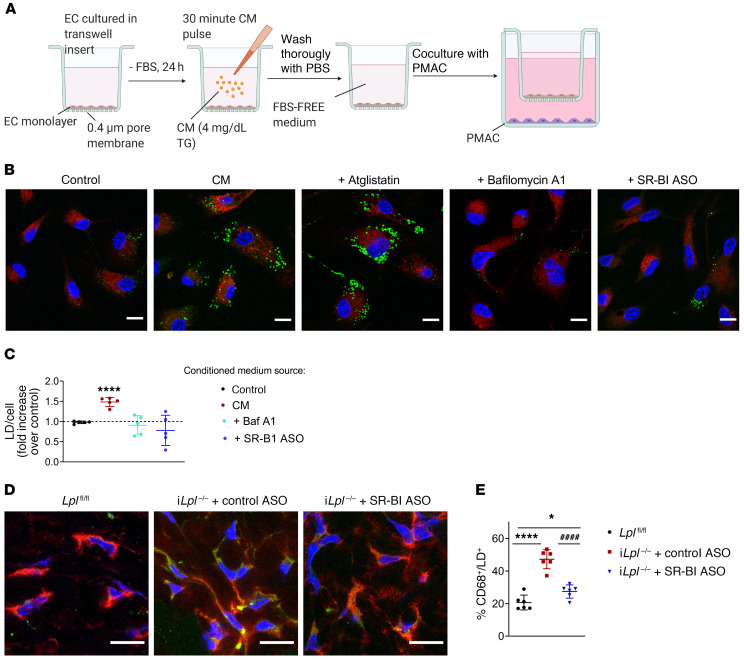
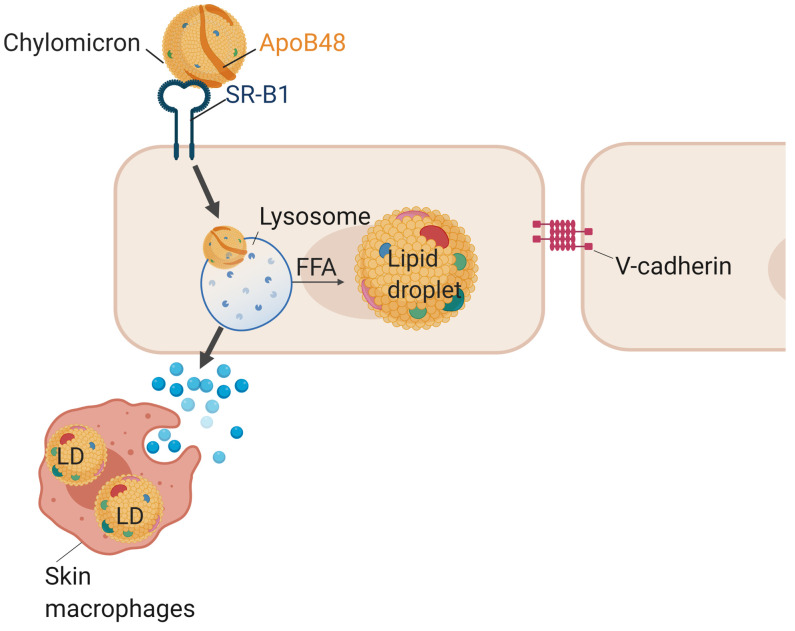
Although tissue uptake of fatty acids from chylomicrons is primarily via lipoprotein lipase (LpL) hydrolysis of triglycerides (TGs), studies of patients with genetic LpL deficiency suggest additional pathways deliver dietary lipids to tissues. Despite an intact endothelial cell (EC) barrier, hyperchylomicronemic patients accumulate chylomicron-derived lipids within skin macrophages, leading to the clinical finding eruptive xanthomas. We explored whether an LpL-independent pathway exists for transfer of circulating lipids across the EC barrier. We found that LpL-deficient mice had a marked increase in aortic EC lipid droplets before and after a fat gavage. Cultured ECs internalized chylomicrons, which were hydrolyzed within lysosomes. The products of this hydrolysis fueled lipid droplet biogenesis in ECs and triggered lipid accumulation in cocultured macrophages. EC chylomicron uptake was inhibited by competition with HDL and knockdown of the scavenger receptor-BI (SR-BI). In vivo, SR-BI knockdown reduced TG accumulation in aortic ECs and skin macrophages of LpL-deficient mice. Thus, ECs internalize chylomicrons, metabolize them in lysosomes, and either store or release their lipids. This latter process may allow accumulation of TGs within skin macrophages and illustrates a pathway that might be responsible for creation of eruptive xanthomas.
Keywords: Endocrinology; Lipoproteins; Metabolism.
Conflict of interest statement
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
