

Brain-gut axis dysfunction in the pathogenesis of traumatic brain injury
- PMID: 34128471
- PMCID: PMC8203445
- DOI: 10.1172/JCI143777
Brain-gut axis dysfunction in the pathogenesis of traumatic brain injury
Abstract
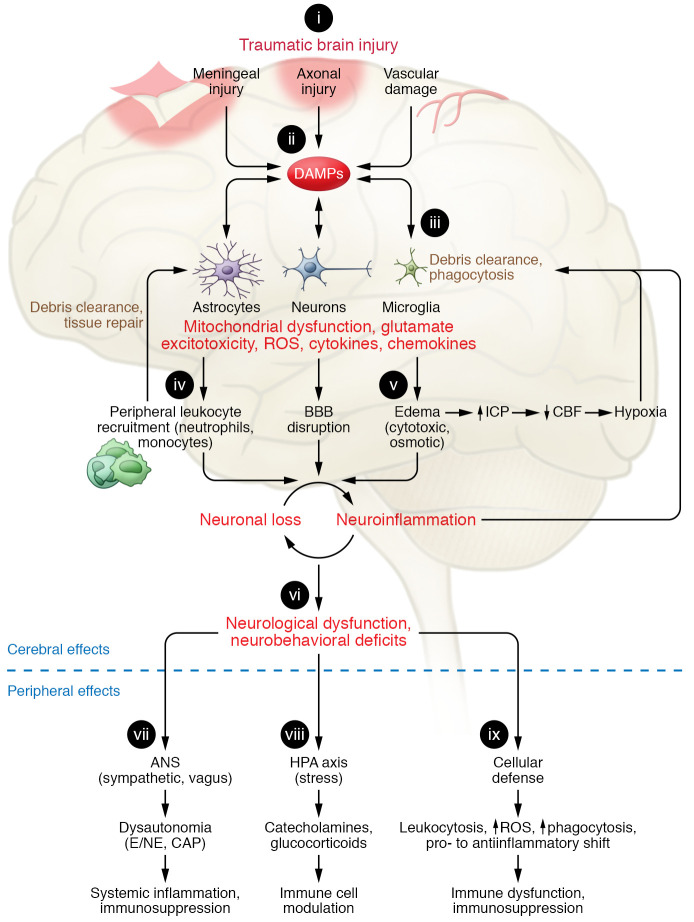
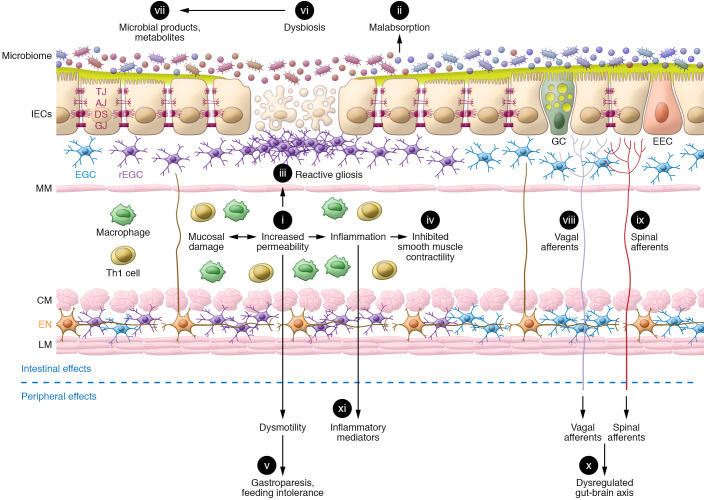
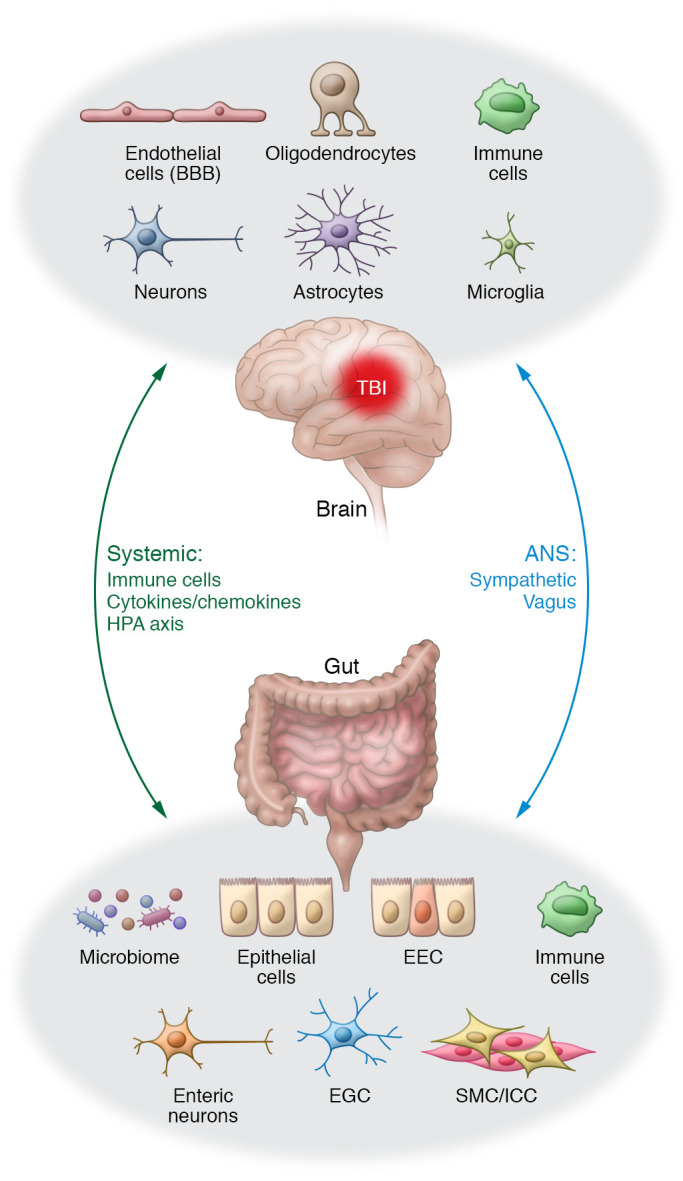
Traumatic brain injury (TBI) is a chronic and progressive disease, and management requires an understanding of both the primary neurological injury and the secondary sequelae that affect peripheral organs, including the gastrointestinal (GI) tract. The brain-gut axis is composed of bidirectional pathways through which TBI-induced neuroinflammation and neurodegeneration impact gut function. The resulting TBI-induced dysautonomia and systemic inflammation contribute to the secondary GI events, including dysmotility and increased mucosal permeability. These effects shape, and are shaped by, changes in microbiota composition and activation of resident and recruited immune cells. Microbial products and immune cell mediators in turn modulate brain-gut activity. Importantly, secondary enteric inflammatory challenges prolong systemic inflammation and worsen TBI-induced neuropathology and neurobehavioral deficits. The importance of brain-gut communication in maintaining GI homeostasis highlights it as a viable therapeutic target for TBI. Currently, treatments directed toward dysautonomia, dysbiosis, and/or systemic inflammation offer the most promise.
Conflict of interest statement
Figures
Similar articles
-
Traumatic Brain Injury and Gut Microbiome: The Role of the Gut-Brain Axis in Neurodegenerative Processes.Curr Neurol Neurosci Rep. 2025 Mar 15;25(1):23. doi: 10.1007/s11910-025-01410-0. Curr Neurol Neurosci Rep. 2025. PMID: 40087204 Review.
-
Bidirectional brain-gut interactions and chronic pathological changes after traumatic brain injury in mice.Brain Behav Immun. 2017 Nov;66:56-69. doi: 10.1016/j.bbi.2017.06.018. Epub 2017 Jul 1. Brain Behav Immun. 2017. PMID: 28676351 Free PMC article.
-
The bidirectional gut-brain-microbiota axis as a potential nexus between traumatic brain injury, inflammation, and disease.Brain Behav Immun. 2017 Nov;66:31-44. doi: 10.1016/j.bbi.2017.05.009. Epub 2017 May 17. Brain Behav Immun. 2017. PMID: 28526435 Review.
-
Acute colitis during chronic experimental traumatic brain injury in mice induces dysautonomia and persistent extraintestinal, systemic, and CNS inflammation with exacerbated neurological deficits.J Neuroinflammation. 2021 Jan 18;18(1):24. doi: 10.1186/s12974-020-02067-x. J Neuroinflammation. 2021. PMID: 33461596 Free PMC article.
-
The Gut-Brain Axis and Neuroinflammation in Traumatic Brain Injury.Mol Neurobiol. 2025 Apr;62(4):4576-4590. doi: 10.1007/s12035-024-04585-8. Epub 2024 Oct 28. Mol Neurobiol. 2025. PMID: 39466574 Review.
Cited by
-
Neuroprotection strategies in traumatic brain injury: Studying the effectiveness of different clinical approaches.Surg Neurol Int. 2024 Jan 26;15:29. doi: 10.25259/SNI_773_2023. eCollection 2024. Surg Neurol Int. 2024. PMID: 38344087 Free PMC article. Review.
-
Roles of HMGB1 on life-threatening traumatic brain injury and sequential peripheral organ damage.Sci Rep. 2024 Sep 13;14(1):21421. doi: 10.1038/s41598-024-72318-x. Sci Rep. 2024. PMID: 39271757 Free PMC article.
-
Translocation and Dissemination of Gut Bacteria after Severe Traumatic Brain Injury.Microorganisms. 2022 Oct 21;10(10):2082. doi: 10.3390/microorganisms10102082. Microorganisms. 2022. PMID: 36296362 Free PMC article.
-
Emerging Neuroprotective Strategies: Unraveling the Potential of HDAC Inhibitors in Traumatic Brain Injury Management.Curr Neuropharmacol. 2024;22(14):2298-2313. doi: 10.2174/1570159X22666240128002056. Curr Neuropharmacol. 2024. PMID: 38288835 Free PMC article. Review.
-
The Intestinal Microbiome after Traumatic Injury.Microorganisms. 2023 Aug 2;11(8):1990. doi: 10.3390/microorganisms11081990. Microorganisms. 2023. PMID: 37630549 Free PMC article. Review.
References
-
- Faul M, et al. CDC. Traumatic brain injury in the United States; emergency department visits, hospitalizations, and deaths, 2002–2006. https://stacks.cdc.gov/view/cdc/5571 Updated March 2010. Accessed August 10, 2020.
