

Stabilization of fatty acid synthesis enzyme acetyl-CoA carboxylase 1 suppresses acute myeloid leukemia development
- PMID: 34128473
- PMCID: PMC8203453
- DOI: 10.1172/JCI141529
Stabilization of fatty acid synthesis enzyme acetyl-CoA carboxylase 1 suppresses acute myeloid leukemia development
Abstract
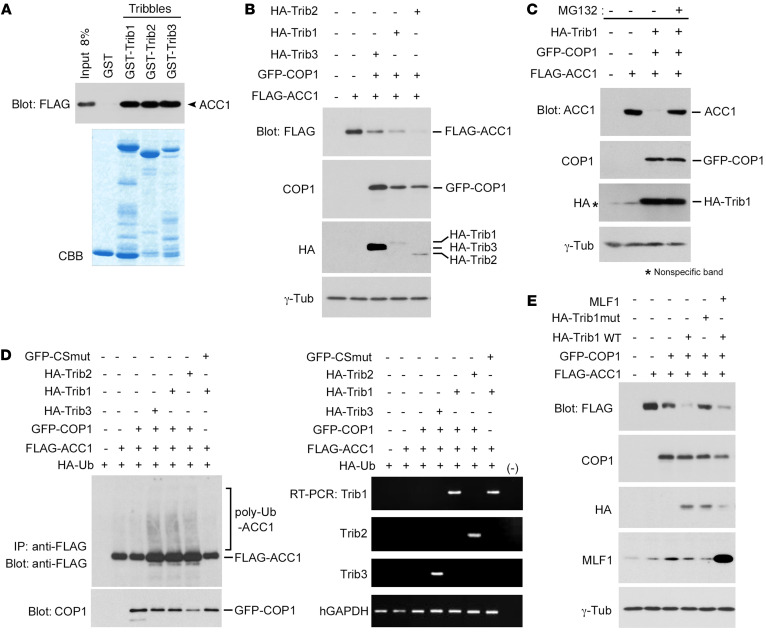
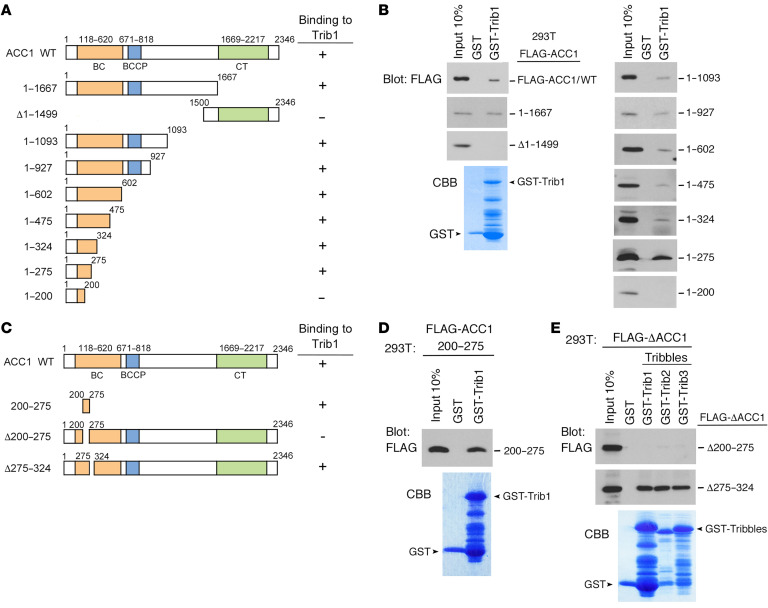
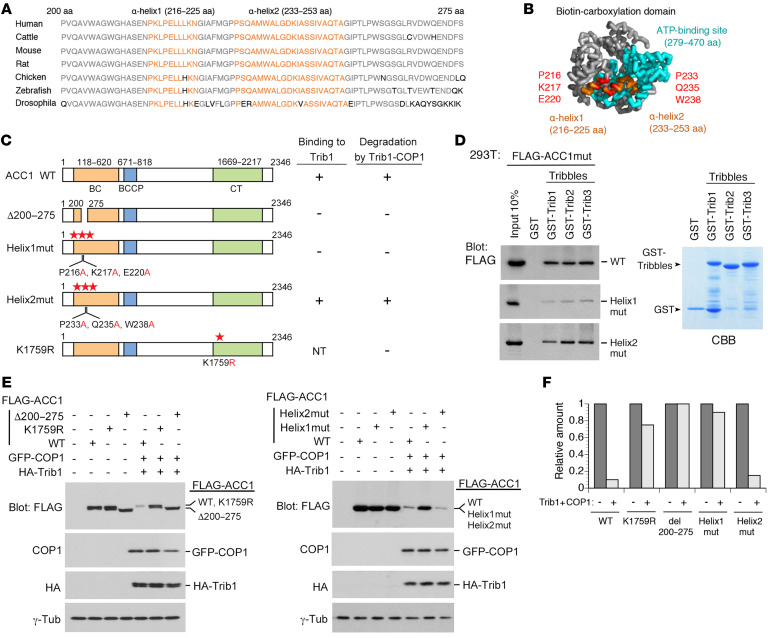
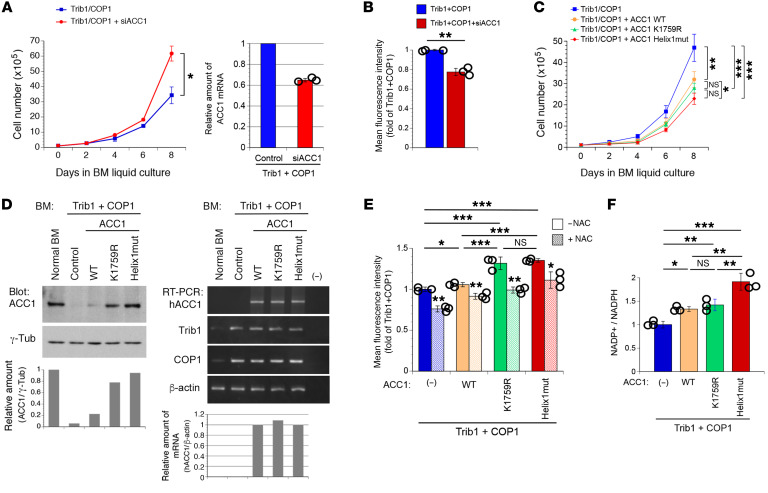
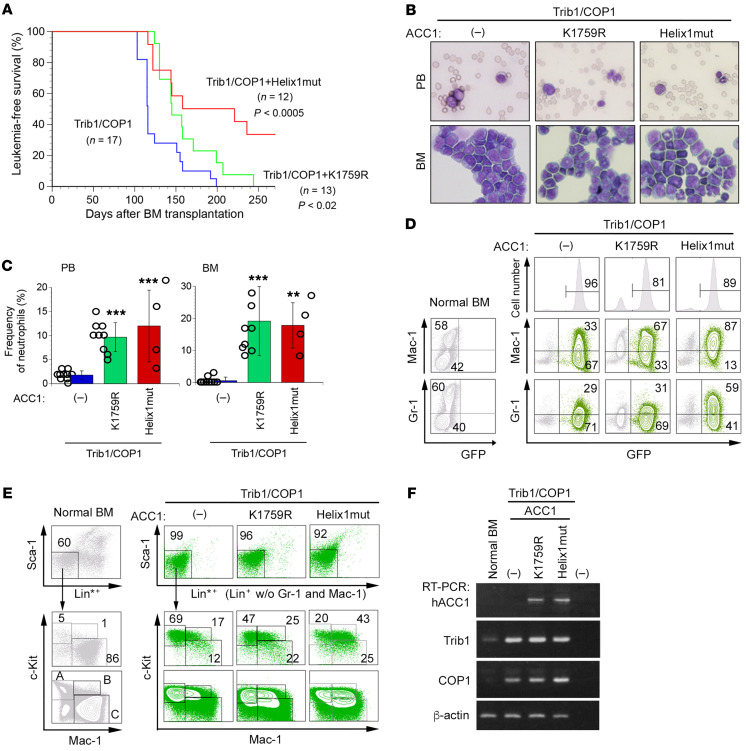
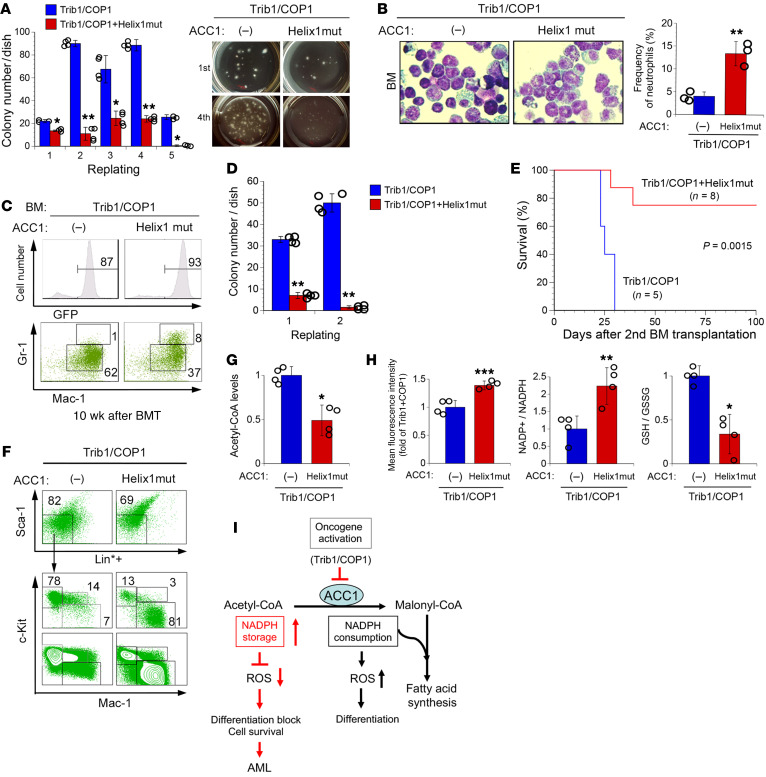
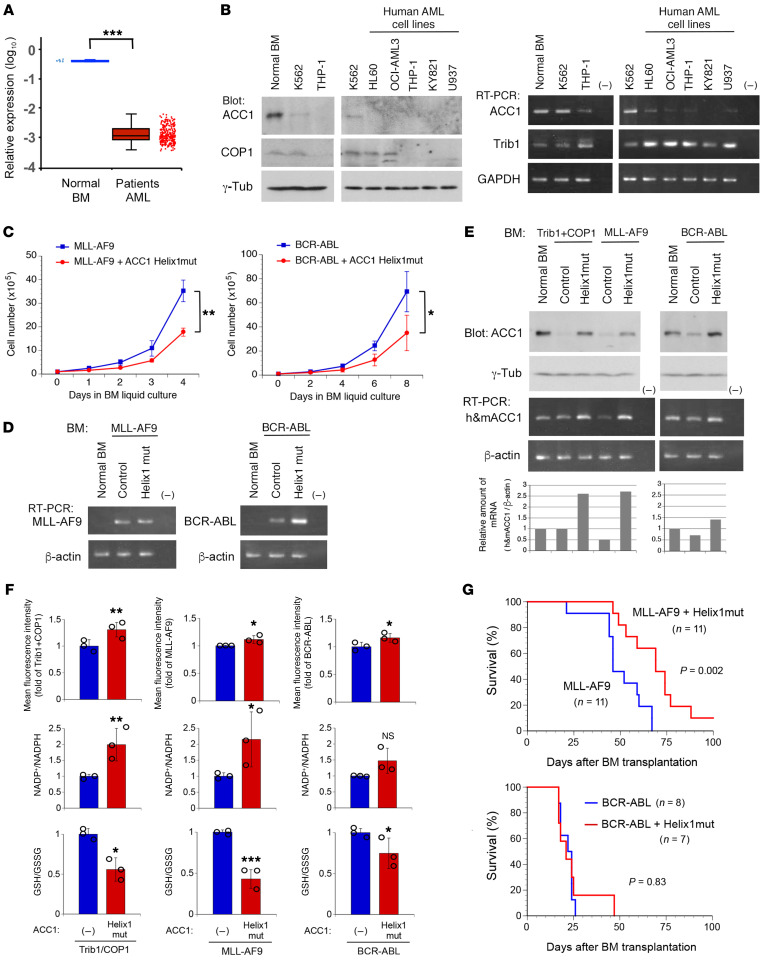
Cancer cells reprogram lipid metabolism during their malignant progression, but limited information is currently available on the involvement of alterations in fatty acid synthesis in cancer development. We herein demonstrate that acetyl-CoA carboxylase 1 (ACC1), a rate-limiting enzyme for fatty acid synthesis, plays a critical role in regulating the growth and differentiation of leukemia-initiating cells. The Trib1-COP1 complex is an E3 ubiquitin ligase that targets C/EBPA, a transcription factor regulating myeloid differentiation, for degradation, and its overexpression specifically induces acute myeloid leukemia (AML). We identified ACC1 as a target of the Trib1-COP1 complex and found that an ACC1 mutant resistant to degradation because of the lack of a Trib1-binding site attenuated complex-driven leukemogenesis. Stable ACC1 protein expression suppressed the growth-promoting activity and increased ROS levels with the consumption of NADPH in a primary bone marrow culture, and delayed the onset of AML with increases in mature myeloid cells in mouse models. ACC1 promoted the terminal differentiation of Trib1-COP1-expressing cells and eradicated leukemia-initiating cells in the early phase of leukemic progression. These results indicate that ACC1 is a natural inhibitor of AML development. The upregulated expression of the ACC1 protein has potential as an effective strategy for cancer therapy.
Keywords: Cancer; Oncology; Tumor suppressors; Ubiquitin-proteosome system.
Conflict of interest statement
Figures
Similar articles
-
Myeloid leukemia factor 1 stabilizes tumor suppressor C/EBPα to prevent Trib1-driven acute myeloid leukemia.Blood Adv. 2017 Sep 1;1(20):1682-1693. doi: 10.1182/bloodadvances.2017007054. eCollection 2017 Sep 12. Blood Adv. 2017. PMID: 29296815 Free PMC article.
-
COP1 targets C/EBPα for degradation and induces acute myeloid leukemia via Trib1.Blood. 2013 Sep 5;122(10):1750-60. doi: 10.1182/blood-2012-12-476101. Epub 2013 Jul 24. Blood. 2013. PMID: 23884858
-
Rapid increase of C/EBPα p42 induces growth arrest of acute myeloid leukemia (AML) cells by Cop1 deletion in Trib1-expressing AML.Leukemia. 2024 Dec;38(12):2585-2597. doi: 10.1038/s41375-024-02430-4. Epub 2024 Oct 4. Leukemia. 2024. PMID: 39367171
-
Role of reversible phosphorylation of acetyl-CoA carboxylase in long-chain fatty acid synthesis.FASEB J. 1989 Sep;3(11):2250-6. doi: 10.1096/fasebj.3.11.2570725. FASEB J. 1989. PMID: 2570725 Review.
-
Acetyl-CoA carboxylase (ACC) as a therapeutic target for metabolic syndrome and recent developments in ACC1/2 inhibitors.Expert Opin Investig Drugs. 2019 Oct;28(10):917-930. doi: 10.1080/13543784.2019.1657825. Epub 2019 Aug 29. Expert Opin Investig Drugs. 2019. PMID: 31430206 Review.
Cited by
-
Lipid and glucose metabolism in senescence.Front Nutr. 2023 Aug 23;10:1157352. doi: 10.3389/fnut.2023.1157352. eCollection 2023. Front Nutr. 2023. PMID: 37680899 Free PMC article. Review.
-
Targeting dysregulated lipid metabolism in the tumor microenvironment.Arch Pharm Res. 2023 Dec;46(11-12):855-881. doi: 10.1007/s12272-023-01473-y. Epub 2023 Dec 7. Arch Pharm Res. 2023. PMID: 38060103 Free PMC article. Review.
-
m6A-Modified circTET2 Interacting with HNRNPC Regulates Fatty Acid Oxidation to Promote the Proliferation of Chronic Lymphocytic Leukemia.Adv Sci (Weinh). 2023 Dec;10(34):e2304895. doi: 10.1002/advs.202304895. Epub 2023 Oct 11. Adv Sci (Weinh). 2023. PMID: 37821382 Free PMC article.
-
Metabolic targeting of platelets to combat thrombosis: dawn of a new paradigm?Cardiovasc Res. 2023 Nov 25;119(15):2497-2507. doi: 10.1093/cvr/cvad149. Cardiovasc Res. 2023. PMID: 37706546 Free PMC article. Review.
-
Targeting Dysregulated Lipid Metabolism in Cancer with Pharmacological Inhibitors.Cancers (Basel). 2024 Mar 28;16(7):1313. doi: 10.3390/cancers16071313. Cancers (Basel). 2024. PMID: 38610991 Free PMC article. Review.
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
