

Multi-omic profiling reveals widespread dysregulation of innate immunity and hematopoiesis in COVID-19
- PMID: 34128959
- PMCID: PMC8210586
- DOI: 10.1084/jem.20210582
Multi-omic profiling reveals widespread dysregulation of innate immunity and hematopoiesis in COVID-19
Abstract
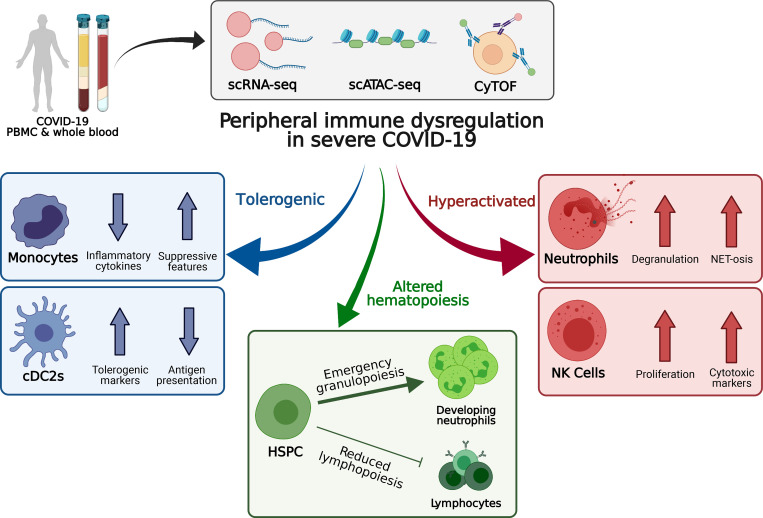
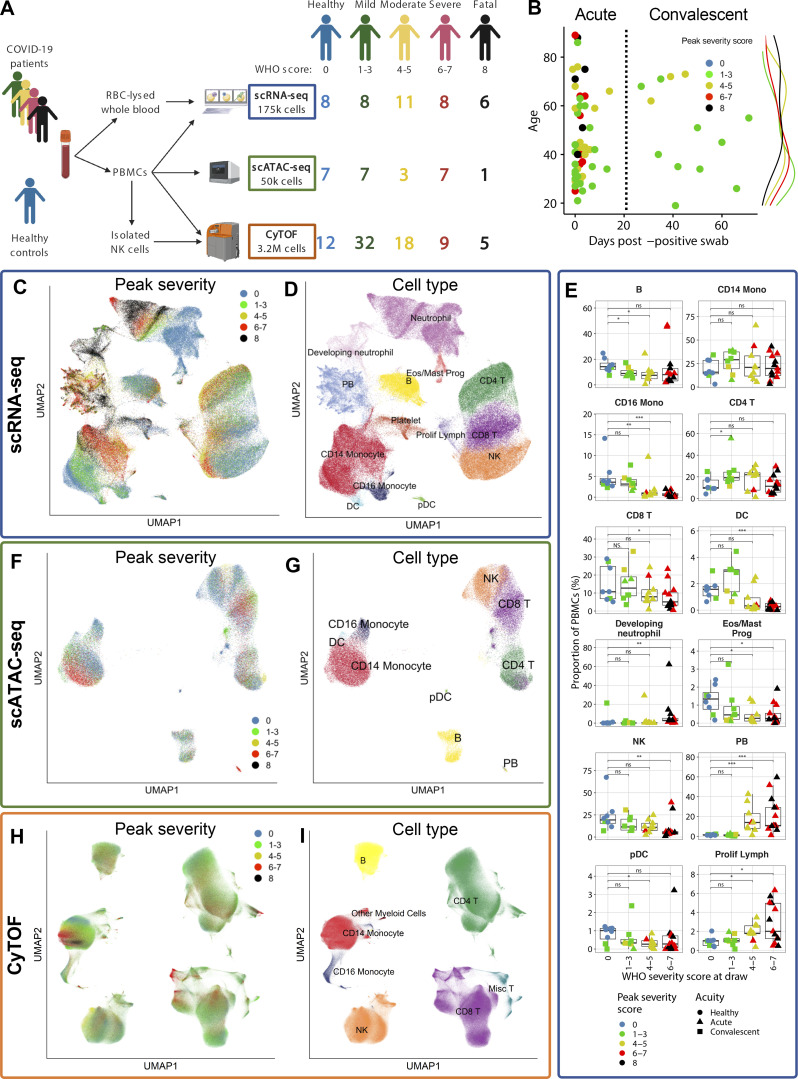
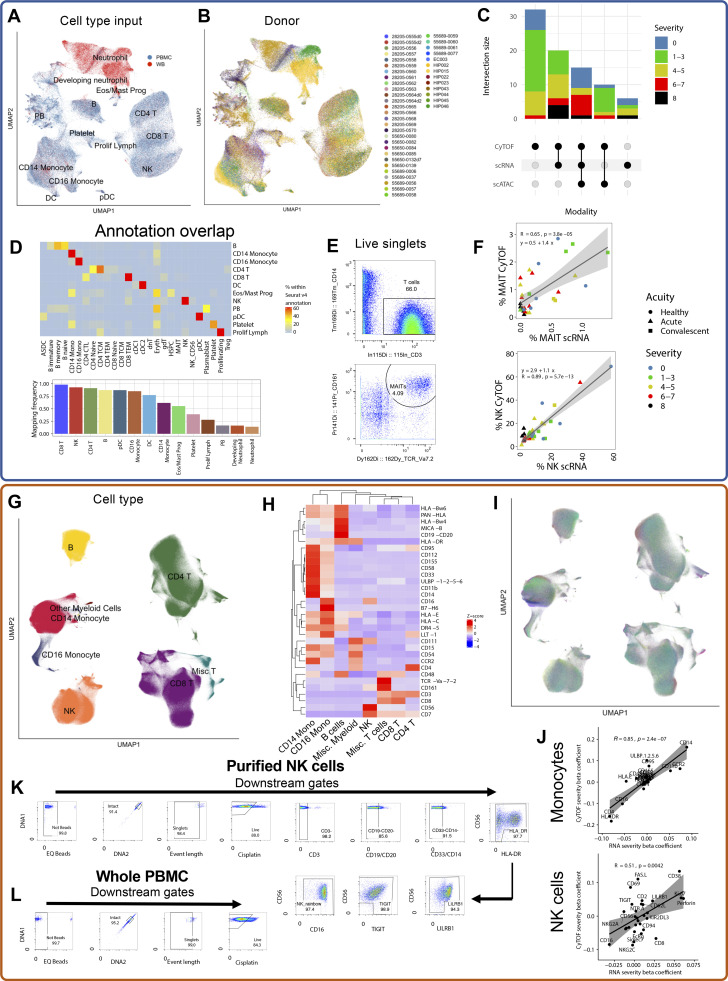
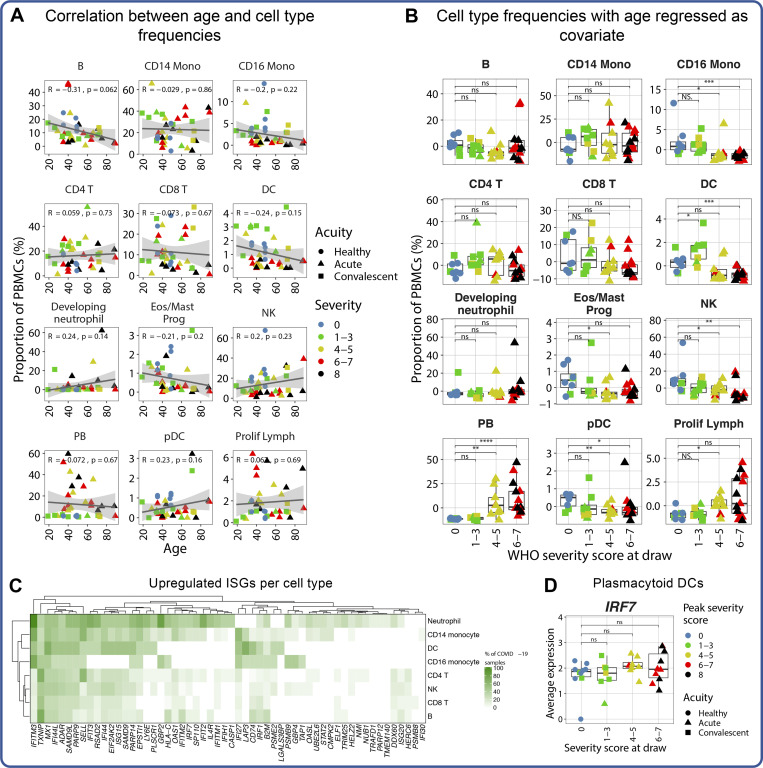
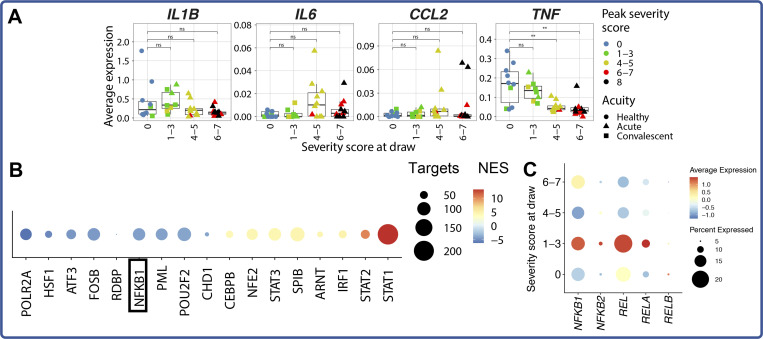
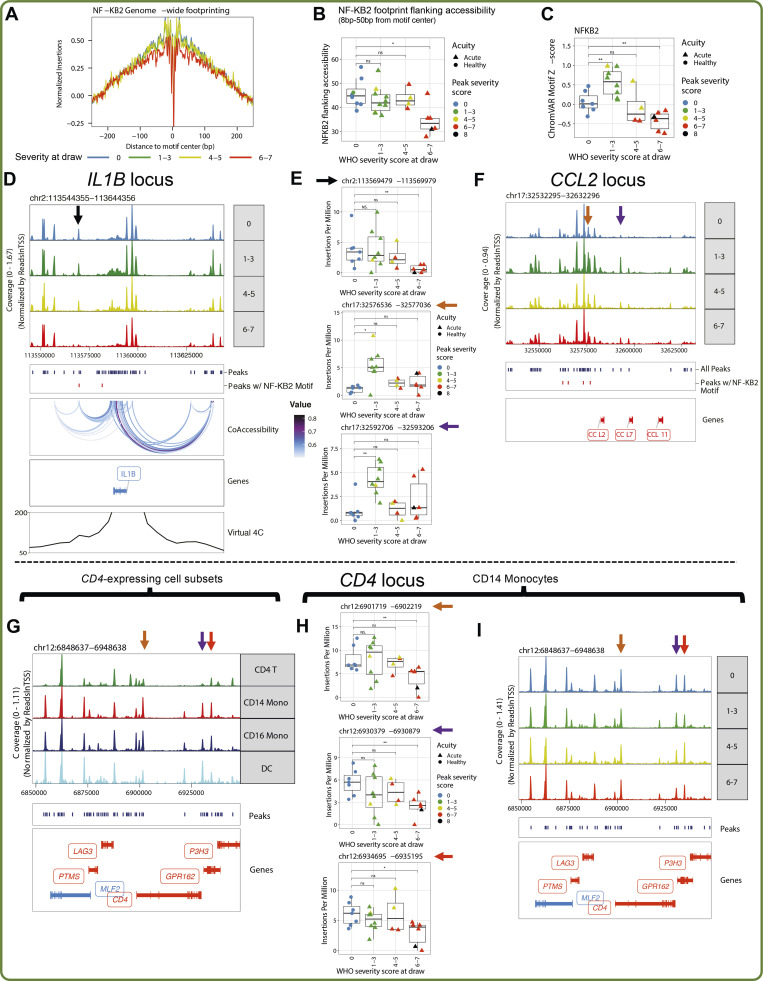
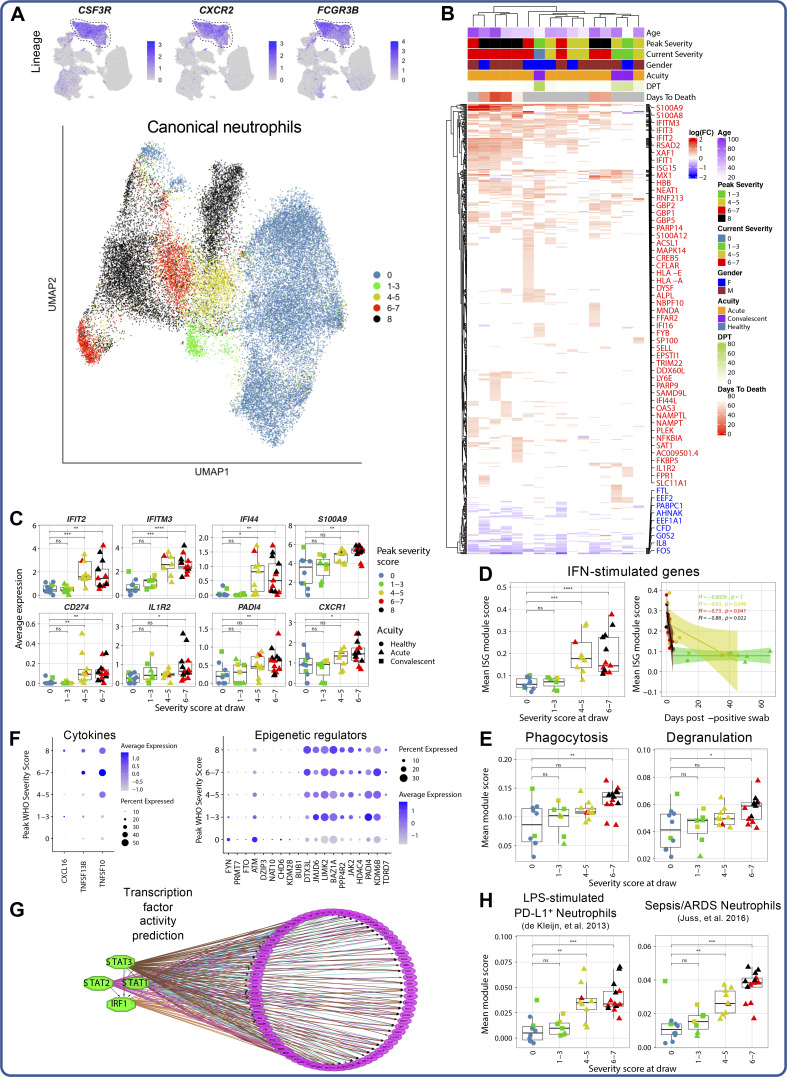
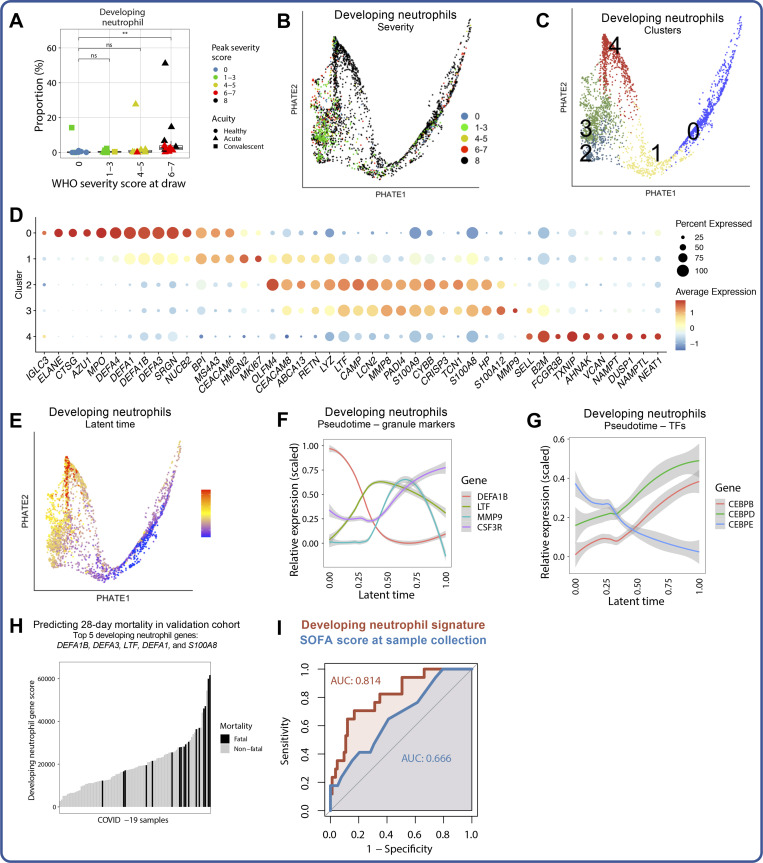
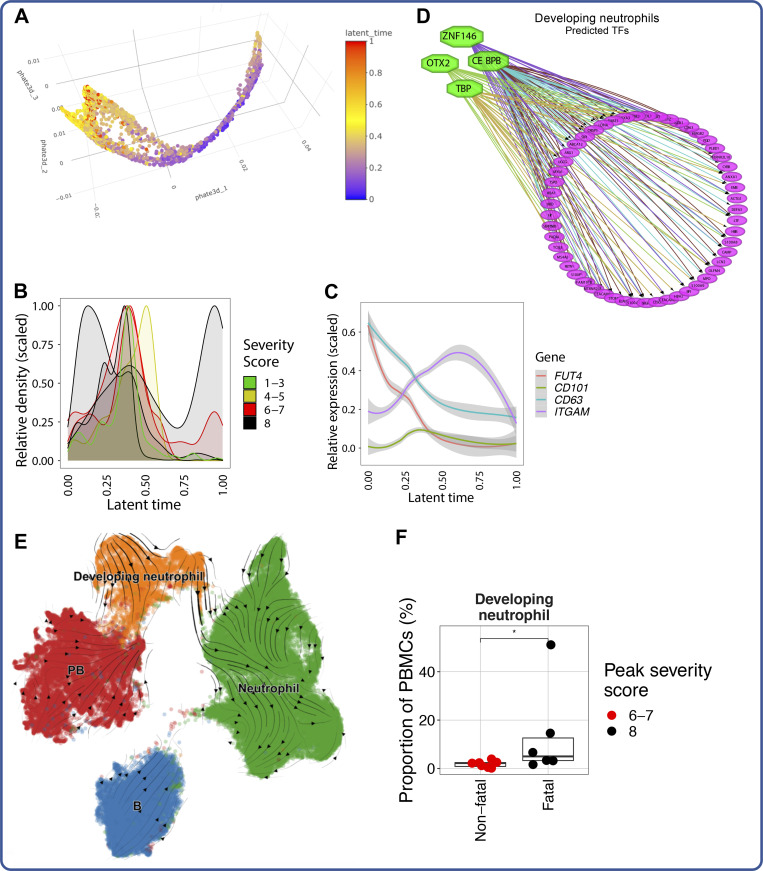
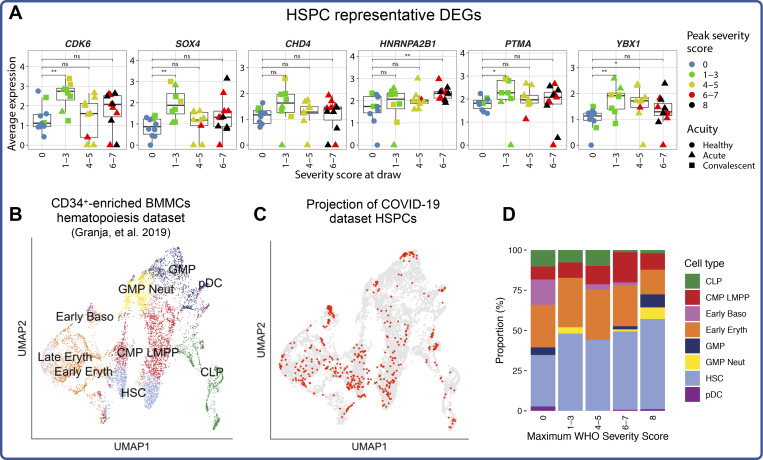
Our understanding of protective versus pathological immune responses to SARS-CoV-2, the virus that causes coronavirus disease 2019 (COVID-19), is limited by inadequate profiling of patients at the extremes of the disease severity spectrum. Here, we performed multi-omic single-cell immune profiling of 64 COVID-19 patients across the full range of disease severity, from outpatients with mild disease to fatal cases. Our transcriptomic, epigenomic, and proteomic analyses revealed widespread dysfunction of peripheral innate immunity in severe and fatal COVID-19, including prominent hyperactivation signatures in neutrophils and NK cells. We also identified chromatin accessibility changes at NF-κB binding sites within cytokine gene loci as a potential mechanism for the striking lack of pro-inflammatory cytokine production observed in monocytes in severe and fatal COVID-19. We further demonstrated that emergency myelopoiesis is a prominent feature of fatal COVID-19. Collectively, our results reveal disease severity-associated immune phenotypes in COVID-19 and identify pathogenesis-associated pathways that are potential targets for therapeutic intervention.
© 2021 Wilk et al.
Conflict of interest statement
Disclosures: A.J. Wilk reported grants from Stanford University Interdisciplinary Graduate Fellowship and NIH during the conduct of the study. M.J. Lee reported grants from NIH during the conduct of the study. B. Wei reported "Stanford University." E.A. Ashley reported "other" from Personalis, Inc., DeepCell, Inc., SVEXA Inc., Astra Zeneca, Gilead, MyoKardia, Amgen, Takeda, Novartis, Genome Medical, Avive, Samsung, Apple Inc., Google, Verily, Disney Inc., Illumina Inc., PacBio, Nanopore, Foresite Capital, and Sequence Bio outside the submitted work. K.C. Nadeau reported grants from National Institute of Allergy and Infectious Diseases, National Heart, Lung, and Blood Institute, Food Allergy Research and Education, and World Allergy Organization; "other" from Cour Pharma, Before Brands, Alladapt, Latitude, IgGenix, Immune Tolerance Network, and National Institutes of Health clinical research centers outside the submitted work; in addition, K.C. Nadeau had a patent to "mixed allergen composition and methods for using the same with royalties paid (Alladapt and Before Brands), a patent to "granulocyte-based methods for detecting and monitoring immune system disorders" issued, and a patent to "methods and assays for detecting and quantifying pure subpopulations of white blood cells in immune system disorders" issued. A.J. Rogers reported personal fees from Merck outside the submitted work. W.J. Greenleaf reported personal fees from 10x Genomics during the conduct of the study, and personal fees from Guardant Health and Protillion Biosciences outside the submitted work; in addition, W.J. Greenleaf had a patent to US20160060691A1 with royalties paid (10x Genomics). C.A. Blish reported personal fees from Catamaran Bio outside the submitted work. No other disclosures were reported.
Figures
References
-
- Alshetaiwi, H., Pervolarakis N., McIntyre L.L., Ma D., Nguyen Q., Rath J.A., Nee K., Hernandez G., Evans K., Torosian L., et al. 2020. Defining the emergence of myeloid-derived suppressor cells in breast cancer using single-cell transcriptomics. Sci. Immunol. 5:eaay6017. 10.1126/sciimmunol.aay6017 - DOI - PMC - PubMed
-
- Arunachalam, P.S., Wimmers F., Mok C.K.P., Perera R.A.P.M., Scott M., Hagan T., Sigal N., Feng Y., Bristow L., Tak-Yin Tsang O., et al. 2020. Systems biological assessment of immunity to mild versus severe COVID-19 infection in humans. Science. 369:1210–1220. 10.1126/science.abc6261 - DOI - PMC - PubMed
-
- Aschenbrenner, A.C., Mouktaroudi M., Krämer B., Oestreich M., Antonakos N., Nuesch-Germano M., Gkizeli K., Bonaguro L., Reusch N., Baßler K., et al. German COVID-19 Omics Initiative (DeCOI) . 2021. Disease severity-specific neutrophil signatures in blood transcriptomes stratify COVID-19 patients. Genome Med. 13:7. 10.1186/s13073-020-00823-5 - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- RM1 HG007735/HG/NHGRI NIH HHS/United States
- T32 GM007365/GM/NIGMS NIH HHS/United States
- T32 AI007290/AI/NIAID NIH HHS/United States
- U19 AI057266/AI/NIAID NIH HHS/United States
- K23 HL125663/HL/NHLBI NIH HHS/United States
- DP1 DA046089/DA/NIDA NIH HHS/United States
- P01 HL108797/HL/NHLBI NIH HHS/United States
- UM1 HG009436/HG/NHGRI NIH HHS/United States
- UM1 HG009442/HG/NHGRI NIH HHS/United States
- R01 HD103571/HD/NICHD NIH HHS/United States
- R25 GM086262/GM/NIGMS NIH HHS/United States
- P50 HG007735/HG/NHGRI NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
