

Reimagining pilocytic astrocytomas in the context of pediatric low-grade gliomas
- PMID: 34131743
- PMCID: PMC8485452
- DOI: 10.1093/neuonc/noab138
Reimagining pilocytic astrocytomas in the context of pediatric low-grade gliomas
Abstract
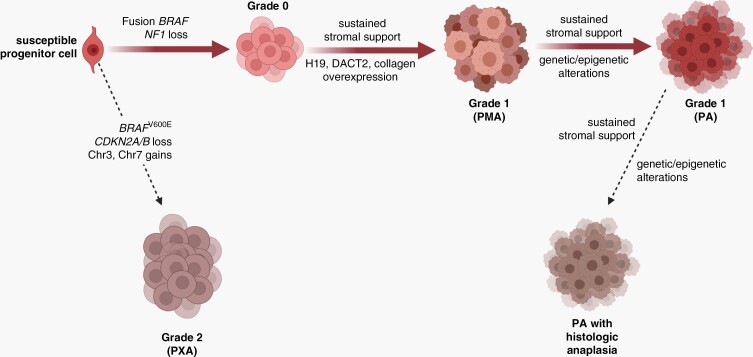
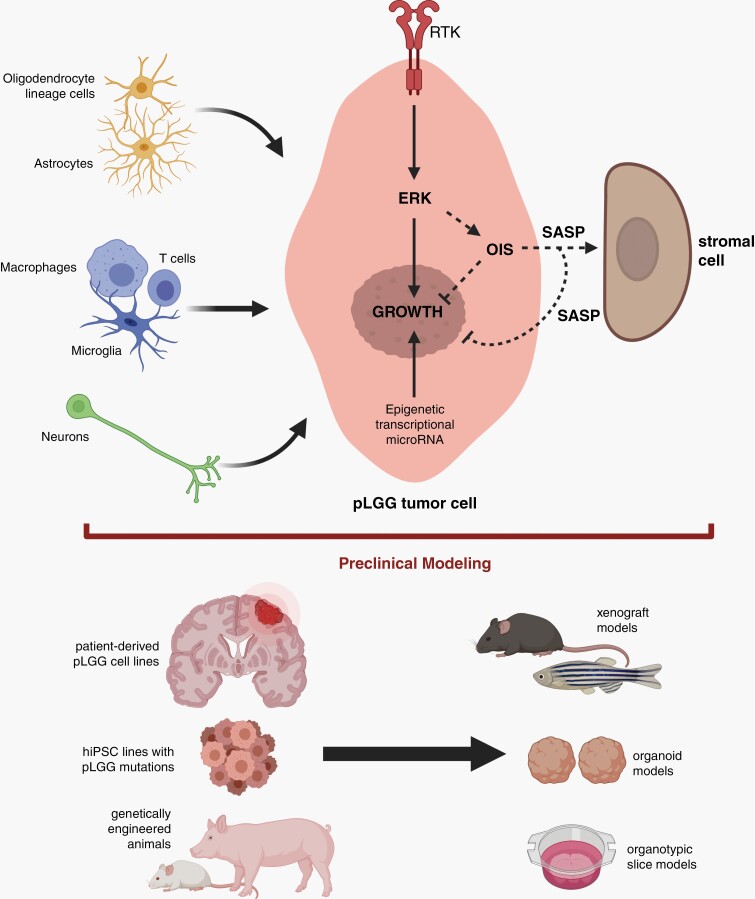
Pediatric low-grade gliomas (pLGGs) are the most common brain tumor in children and are associated with lifelong clinical morbidity. Relative to their high-grade adult counterparts or other malignant childhood brain tumors, there is a paucity of authenticated preclinical models for these pLGGs and an incomplete understanding of their molecular and cellular pathogenesis. While large-scale genomic profiling efforts have identified the majority of pathogenic driver mutations, which converge on the MAPK/ERK signaling pathway, it is now appreciated that these events may not be sufficient by themselves for gliomagenesis and clinical progression. In light of the recent World Health Organization reclassification of pLGGs, and pilocytic astrocytoma (PA), in particular, we review our current understanding of these pediatric brain tumors, provide a conceptual framework for future mechanistic studies, and outline the challenges and pressing needs for the pLGG clinical and research communities.
Keywords: BRAF; MEK; cellular senescence; low-grade glioma; neurofibromatosis type 1; pediatric brain tumor; pilocytic astrocytoma; tumor microenvironment.
© The Author(s) 2021. Published by Oxford University Press on behalf of the Society for Neuro-Oncology. All rights reserved. For permissions, please e-mail: journals.permissions@oup.com.
Figures
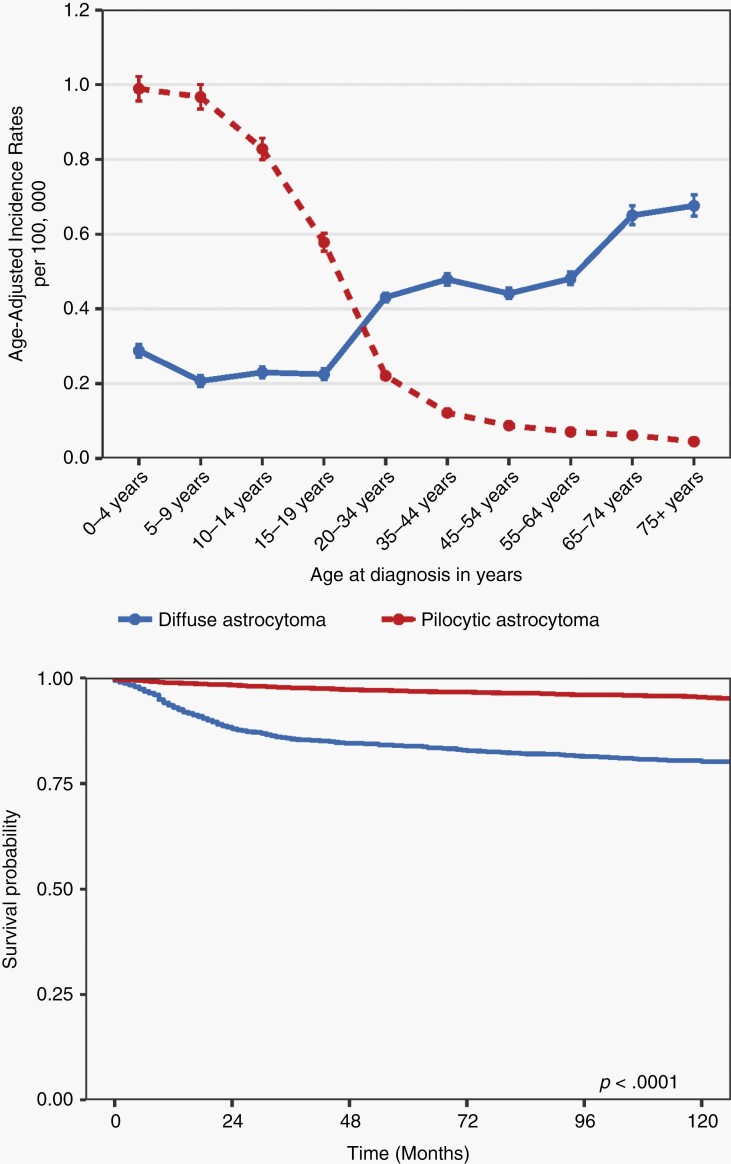
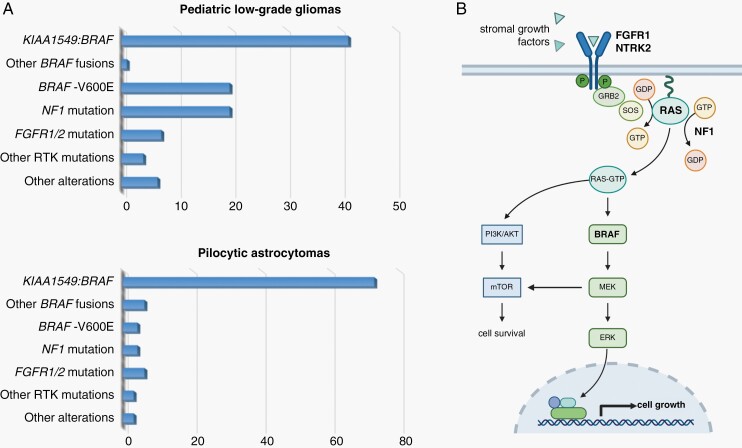
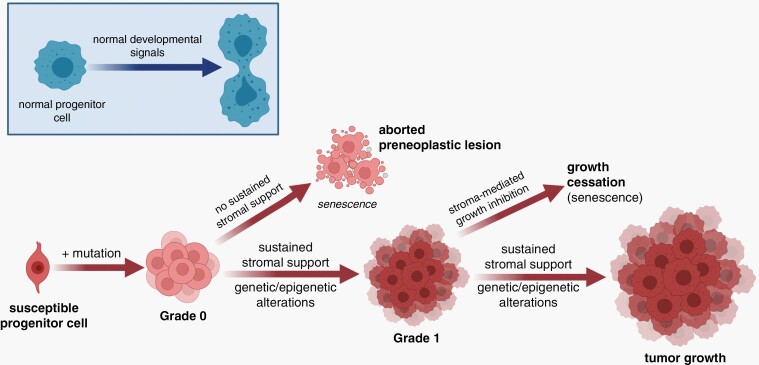
References
-
- Louis DN, Perry A, Reifenberger G, et al. . The 2016 World Health Organization classification of tumors of the central nervous system: a summary. Acta Neuropathol. 2016;131(6):803–820. - PubMed
-
- NPCR and SEER. National Program of Cancer Registries and Surveillance Epidemiology, and End Results Program. SEER*Stat Database: NPCR and SEER Incidence – U.S. Cancer Statistics 2001–2016 Public Use Research Database, 2019 submission (2001–2016), United States Department of Health and Human Services, Centers for Disease Control and Prevention and National Cancer Institute. Released June 2020; Available at www.cdc.gov/cancer/uscs/public-use. Accessed May 18, 2021.
-
- SEER. Surveillance Epidemiology, and End Results Program SEER*Stat Database: Incidence - SEER Research Data, 9 Registries, Nov 2019 Sub (1975-2017) - Linked To County Attributes - Time Dependent (1990-2017) Income/Rurality, 1969-2017 Counties, National Cancer Institute, DCCPS, Surveillance Research Program. Available at www.seer.cancer.gov. Accessed May 18, 2021.
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
