

efam: an expanded, metaproteome-supported HMM profile database of viral protein families
- PMID: 34132786
- PMCID: PMC9502166
- DOI: 10.1093/bioinformatics/btab451
efam: an expanded, metaproteome-supported HMM profile database of viral protein families
Abstract
Motivation: Viruses infect, reprogram and kill microbes, leading to profound ecosystem consequences, from elemental cycling in oceans and soils to microbiome-modulated diseases in plants and animals. Although metagenomic datasets are increasingly available, identifying viruses in them is challenging due to poor representation and annotation of viral sequences in databases.
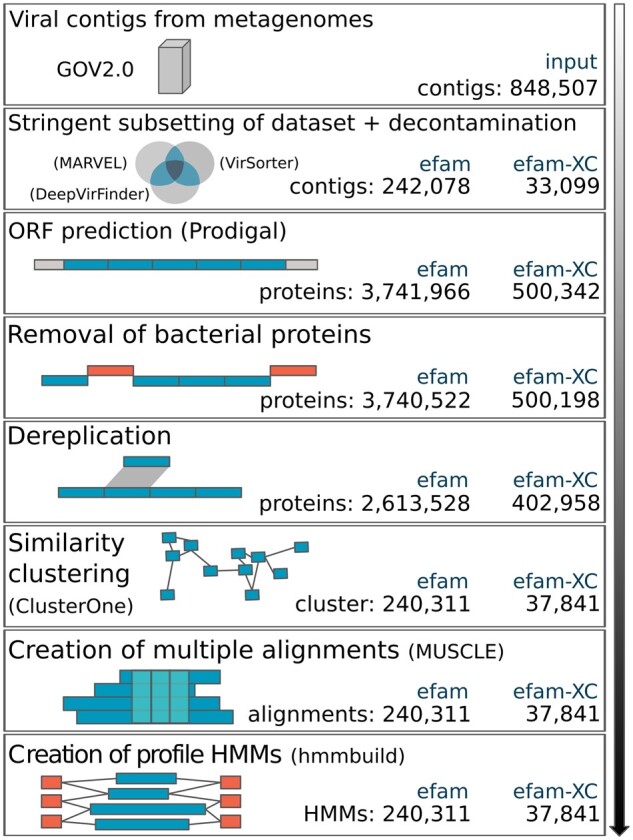
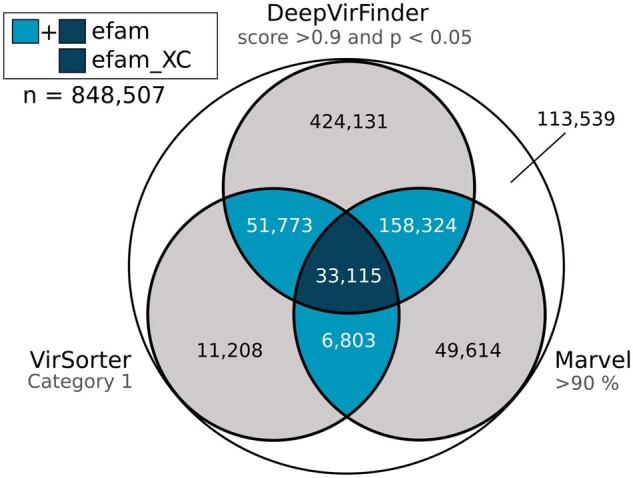
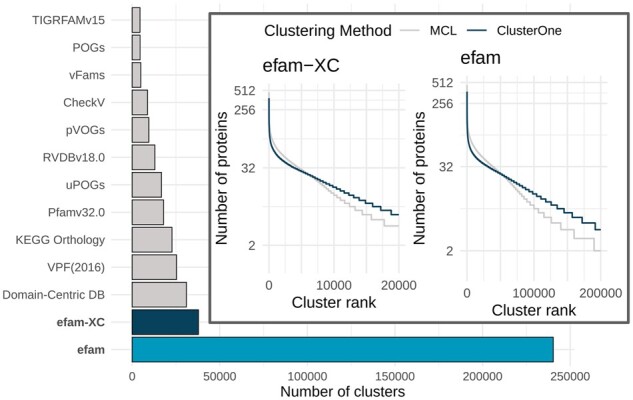
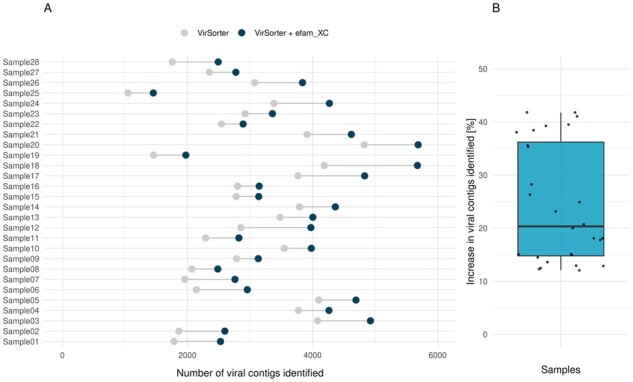
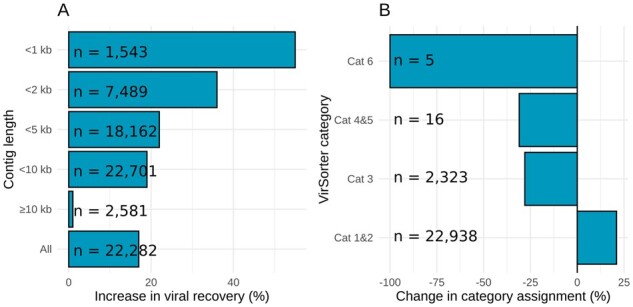
Results: Here, we establish efam, an expanded collection of Hidden Markov Model (HMM) profiles that represent viral protein families conservatively identified from the Global Ocean Virome 2.0 dataset. This resulted in 240 311 HMM profiles, each with at least 2 protein sequences, making efam >7-fold larger than the next largest, pan-ecosystem viral HMM profile database. Adjusting the criteria for viral contig confidence from 'conservative' to 'eXtremely Conservative' resulted in 37 841 HMM profiles in our efam-XC database. To assess the value of this resource, we integrated efam-XC into VirSorter viral discovery software to discover viruses from less-studied, ecologically distinct oxygen minimum zone (OMZ) marine habitats. This expanded database led to an increase in viruses recovered from every tested OMZ virome by ∼24% on average (up to ∼42%) and especially improved the recovery of often-missed shorter contigs (<5 kb). Additionally, to help elucidate lesser-known viral protein functions, we annotated the profiles using multiple databases from the DRAM pipeline and virion-associated metaproteomic data, which doubled the number of annotations obtainable by standard, single-database annotation approaches. Together, these marine resources (efam and efam-XC) are provided as searchable, compressed HMM databases that will be updated bi-annually to help maximize viral sequence discovery and study from any ecosystem.
Availability and implementation: The resources are available on the iVirus platform at (doi.org/10.25739/9vze-4143).
Supplementary information: Supplementary data are available at Bioinformatics online.
© The Author(s) 2021. Published by Oxford University Press.
Figures
References
-
- Boratto P.V.M. et al. (2020) A mysterious 80 nm amoeba virus with a near-complete “ORFan genome” challenges the classification of DNA viruses. bioRxiv, doi: 10.1101/2020.01.28.923185.
