

Case Report: Identification of Novel Variants in ERCC4 and DDB2 Genes in Two Tunisian Patients With Atypical Xeroderma Pigmentosum Phenotype
- PMID: 34135938
- PMCID: PMC8203331
- DOI: 10.3389/fgene.2021.650639
Case Report: Identification of Novel Variants in ERCC4 and DDB2 Genes in Two Tunisian Patients With Atypical Xeroderma Pigmentosum Phenotype
Abstract
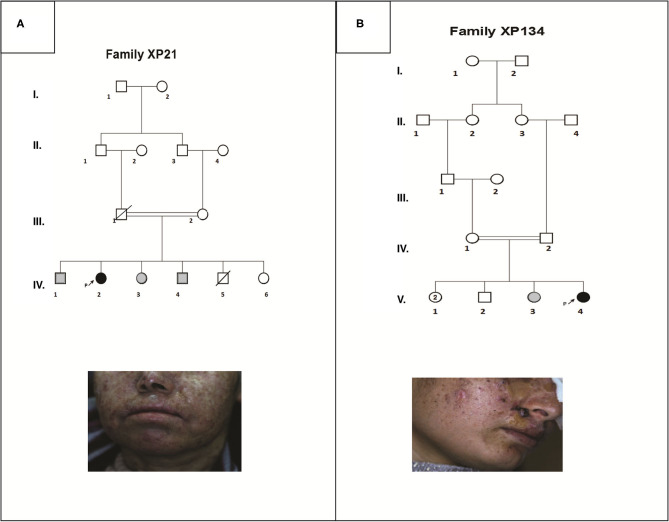
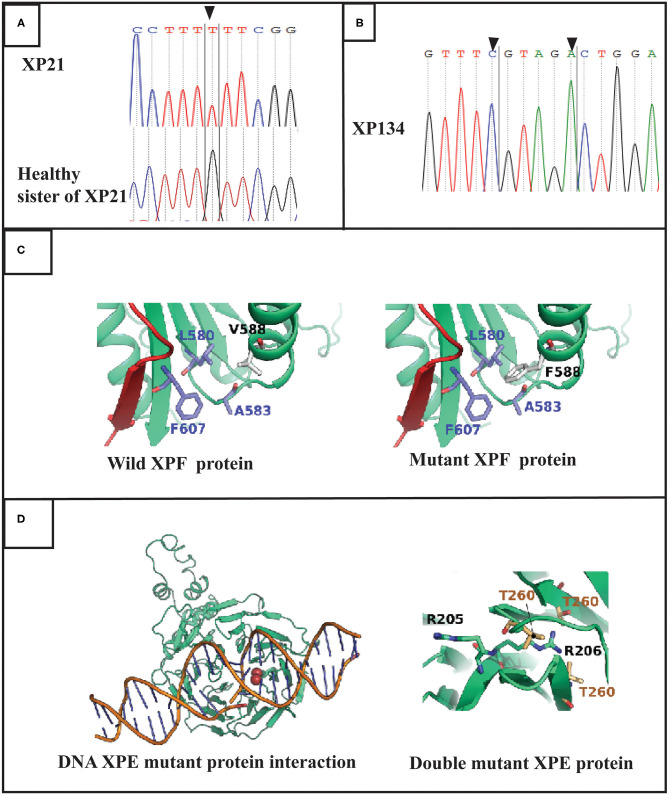
Xeroderma Pigmentosum (XP) is a rare genetic disorder affecting the nucleotide excision repair system (NER). It is characterized by an extreme sensitivity to sunlight that induces cutaneous disorders such as severe sunburn, freckling and cancers. In Tunisia, six complementation groups have been already identified. However, the genetic etiology remains unknown for several patients. In this study, we investigated clinical characteristics and genetic defects in two families with atypical phenotypes originating from the central region in Tunisia. Clinical investigation revealed mild cutaneous features in two patients who develop multiple skin cancers at later ages, with no neurological disorders. Targeted gene sequencing revealed that they carried novel variants. A homozygous variation in the ERCC4 gene c.1762G>T, p.V588F, detected in patient XP21. As for patient XP134, he carried two homozygous mutations in the DDB2 gene c.613T>C, p.C205R and c.618C>A, p.S206R. Structural modeling of the protein predicted the identified ERCC4 variant to mildly affect protein stability without affecting its functional domains. As for the case of DDB2 double mutant, the second variation seems to cause a mild effect on the protein structure unlike the first variation which does not seem to have an effect on it. This study contributes to further characterize the mutation spectrum of XP in Tunisian families. Targeted gene sequencing accelerated the identification of rare unexpected genetic defects for diagnostic testing and genetic counseling.
Keywords: DDB2 gene; ERCC4/XPF; NER defects; skin cancer; xeroderma pigmentosum.
Copyright © 2021 Nabouli, Chikhaoui, Othman, Elouej, Jones, Lagarde, Rekaya, Messaoud, Zghal, Delague, Levy, De Sandre-Giovannoli, Abdelhak and Yacoub-Youssef.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures
Similar articles
-
Identification of a ERCC5 c.2333T>C (L778P) Variant in Two Tunisian Siblings With Mild Xeroderma Pigmentosum Phenotype.Front Genet. 2019 Feb 14;10:111. doi: 10.3389/fgene.2019.00111. eCollection 2019. Front Genet. 2019. PMID: 30838033 Free PMC article.
-
Whole Exome Sequencing allows the identification of two novel groups of Xeroderma pigmentosum in Tunisia, XP-D and XP-E: Impact on molecular diagnosis.J Dermatol Sci. 2018 Feb;89(2):172-180. doi: 10.1016/j.jdermsci.2017.10.015. Epub 2017 Nov 2. J Dermatol Sci. 2018. PMID: 29169765
-
Deep intronic founder mutations identified in the ERCC4/XPF gene are potential therapeutic targets for a high-frequency form of xeroderma pigmentosum.Proc Natl Acad Sci U S A. 2023 Jul 4;120(27):e2217423120. doi: 10.1073/pnas.2217423120. Epub 2023 Jun 26. Proc Natl Acad Sci U S A. 2023. PMID: 37364129 Free PMC article.
-
Identification of a novel DDB2 mutation in a Chinese Han family with Xeroderma pigmentosum group E:a case report and literature review.BMC Med Genet. 2020 Mar 30;21(1):67. doi: 10.1186/s12881-020-00997-0. BMC Med Genet. 2020. PMID: 32228487 Free PMC article. Review.
-
Xeroderma pigmentosum group E and DDB2, a smaller subunit of damage-specific DNA binding protein: proposed classification of xeroderma pigmentosum, Cockayne syndrome, and ultraviolet-sensitive syndrome.J Dermatol Sci. 2006 Feb;41(2):87-96. doi: 10.1016/j.jdermsci.2005.10.010. Epub 2005 Dec 1. J Dermatol Sci. 2006. PMID: 16325378 Review.
Cited by
-
Case report: Variants in the ERCC4 gene as a rare cause of cerebellar ataxia with chorea.Front Genet. 2023 Feb 2;14:1107460. doi: 10.3389/fgene.2023.1107460. eCollection 2023. Front Genet. 2023. PMID: 36816046 Free PMC article.
-
Unveiling Secondary Mutations in Blended Phenotypes: Dual ERCC4 and OTOA Pathogenic Variants Through WES Analysis.Int J Mol Sci. 2024 Dec 16;25(24):13471. doi: 10.3390/ijms252413471. Int J Mol Sci. 2024. PMID: 39769235 Free PMC article.
References
-
- Ben Rekaya M., Naouali C., Messaoud O., Jones M., Bouyacoub Y., Nagara M., et al. . (2017). Whole Exome Sequencing allows the identification of two novel groups of Xeroderma pigmentosum in Tunisia, XP-D and XP-E: impact on molecular diagnosis. J. Dermatol. Sci. 89, 172–180. 10.1016/j.jdermsci.2017.10.015 - DOI - PubMed
Publication types
LinkOut - more resources
Full Text Sources
Research Materials