

Curation Guidelines for de novo Generated Transposable Element Families
- PMID: 34138525
- PMCID: PMC9191830
- DOI: 10.1002/cpz1.154
Curation Guidelines for de novo Generated Transposable Element Families
Abstract
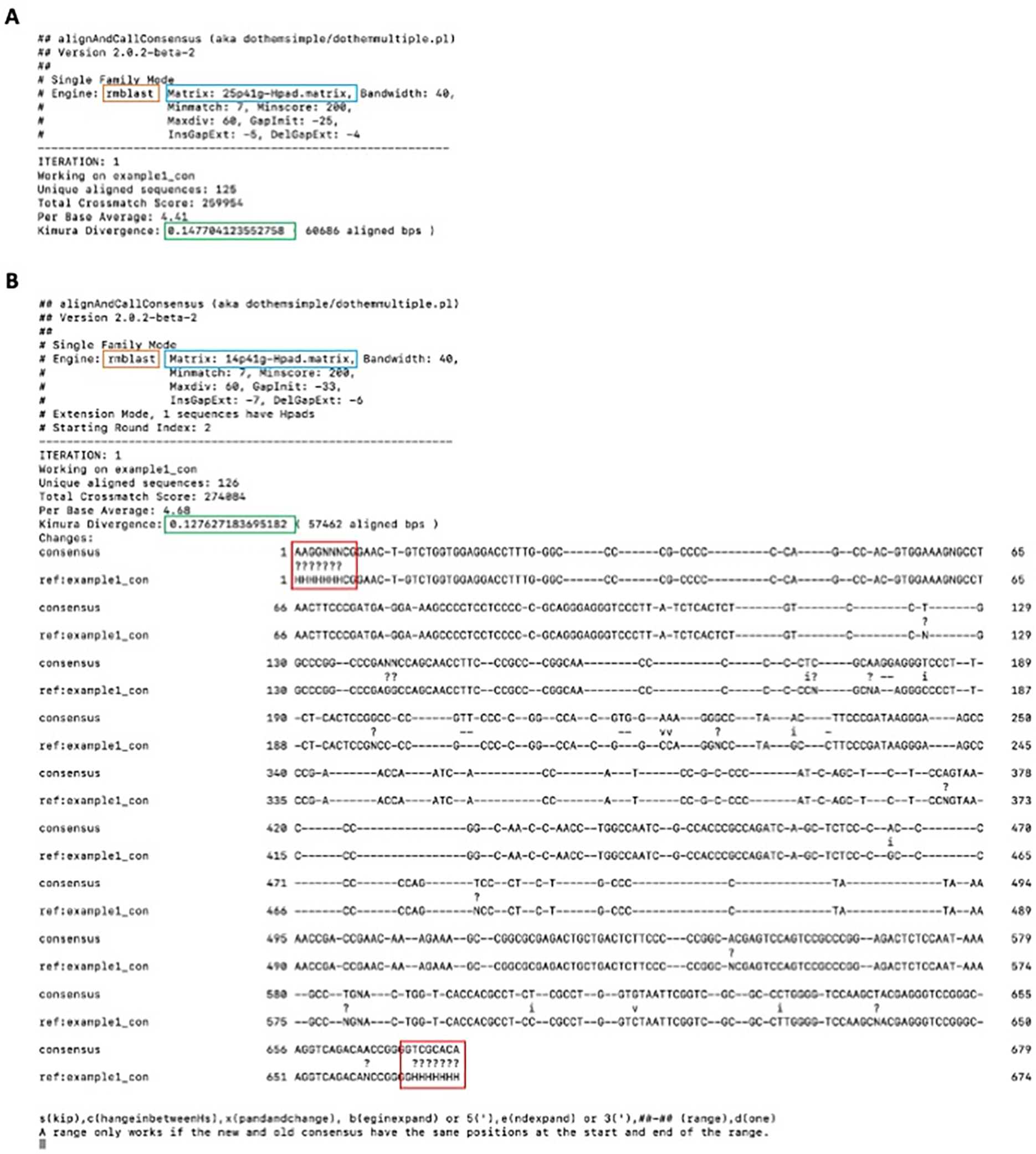
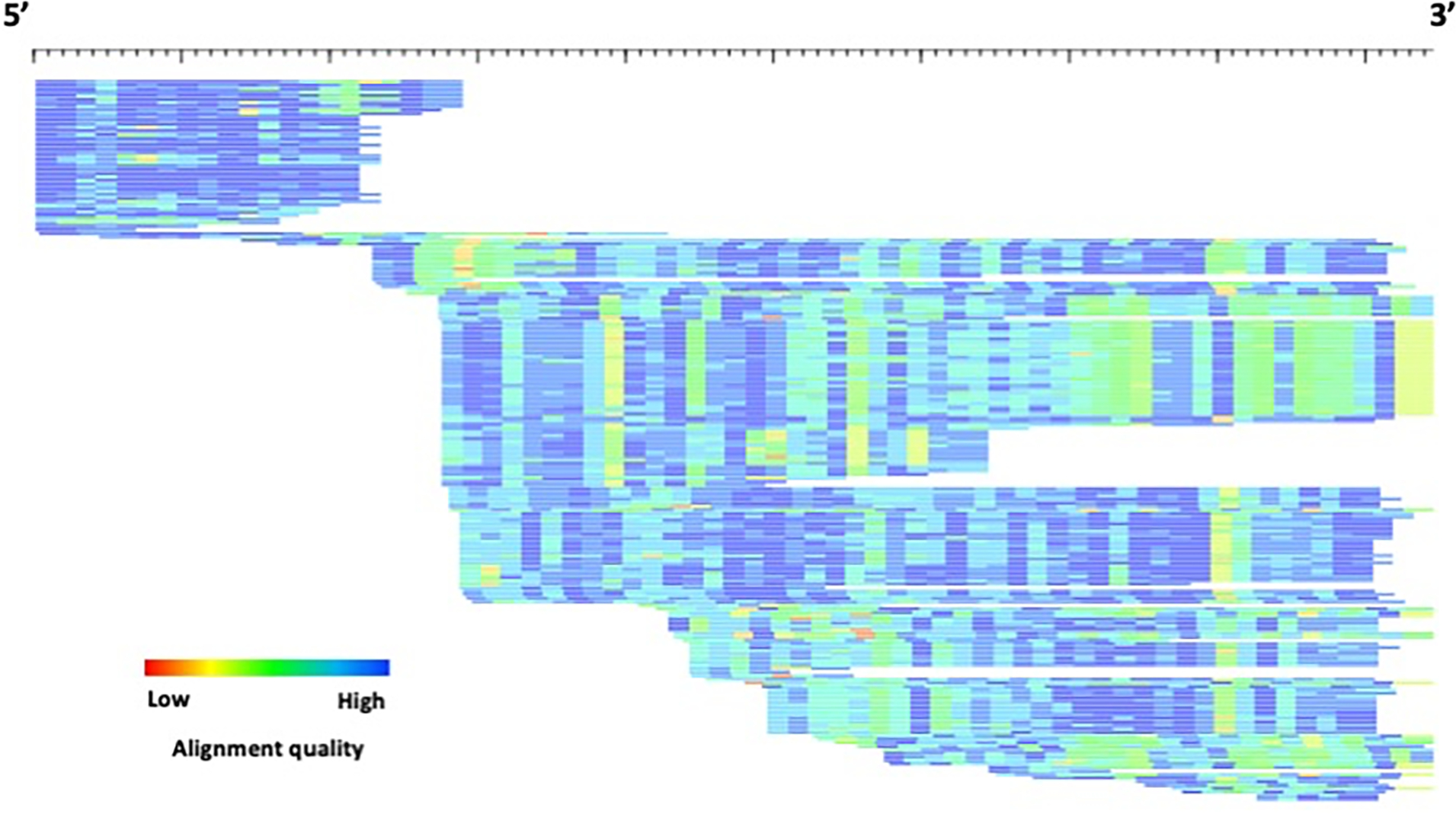
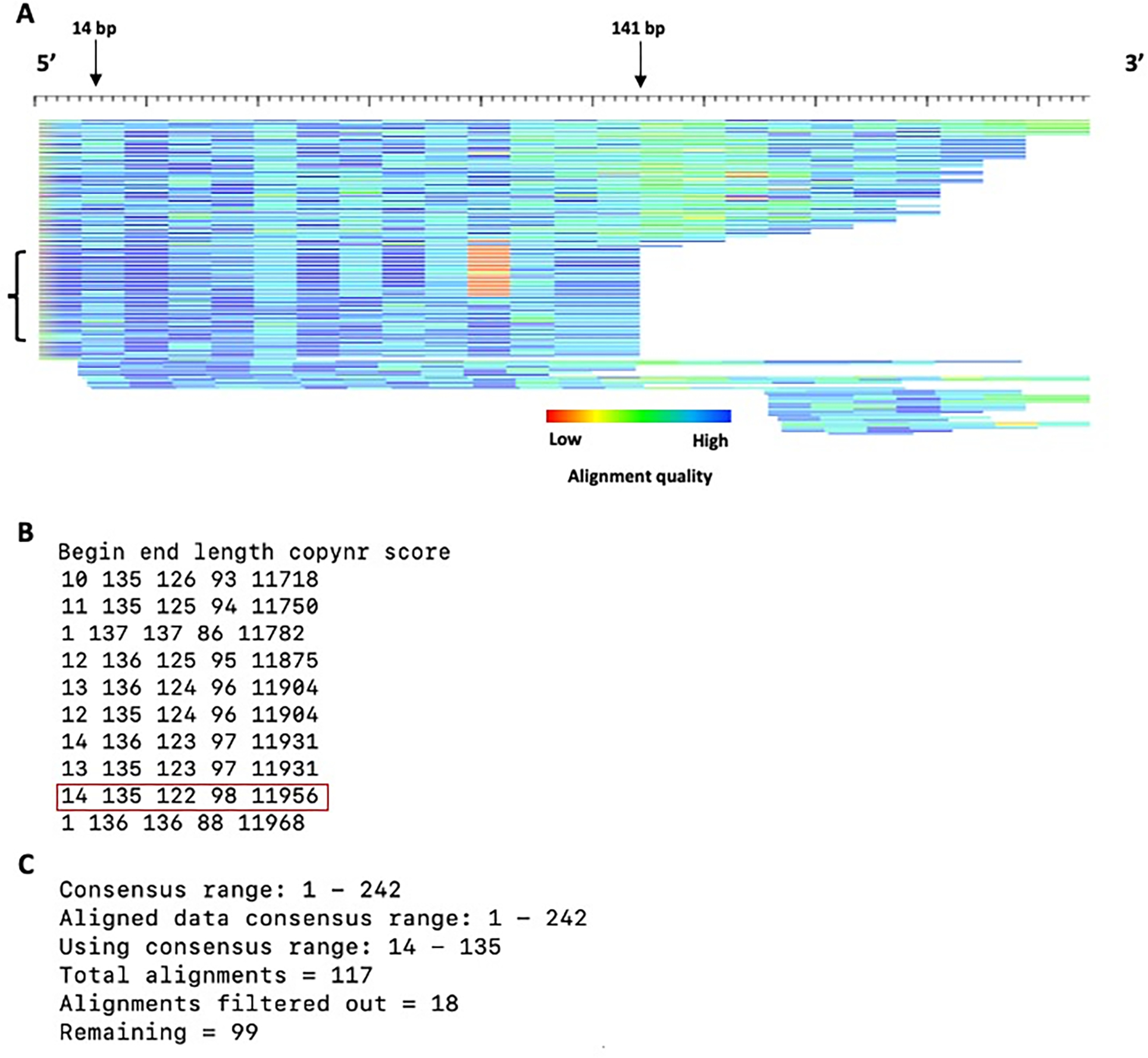
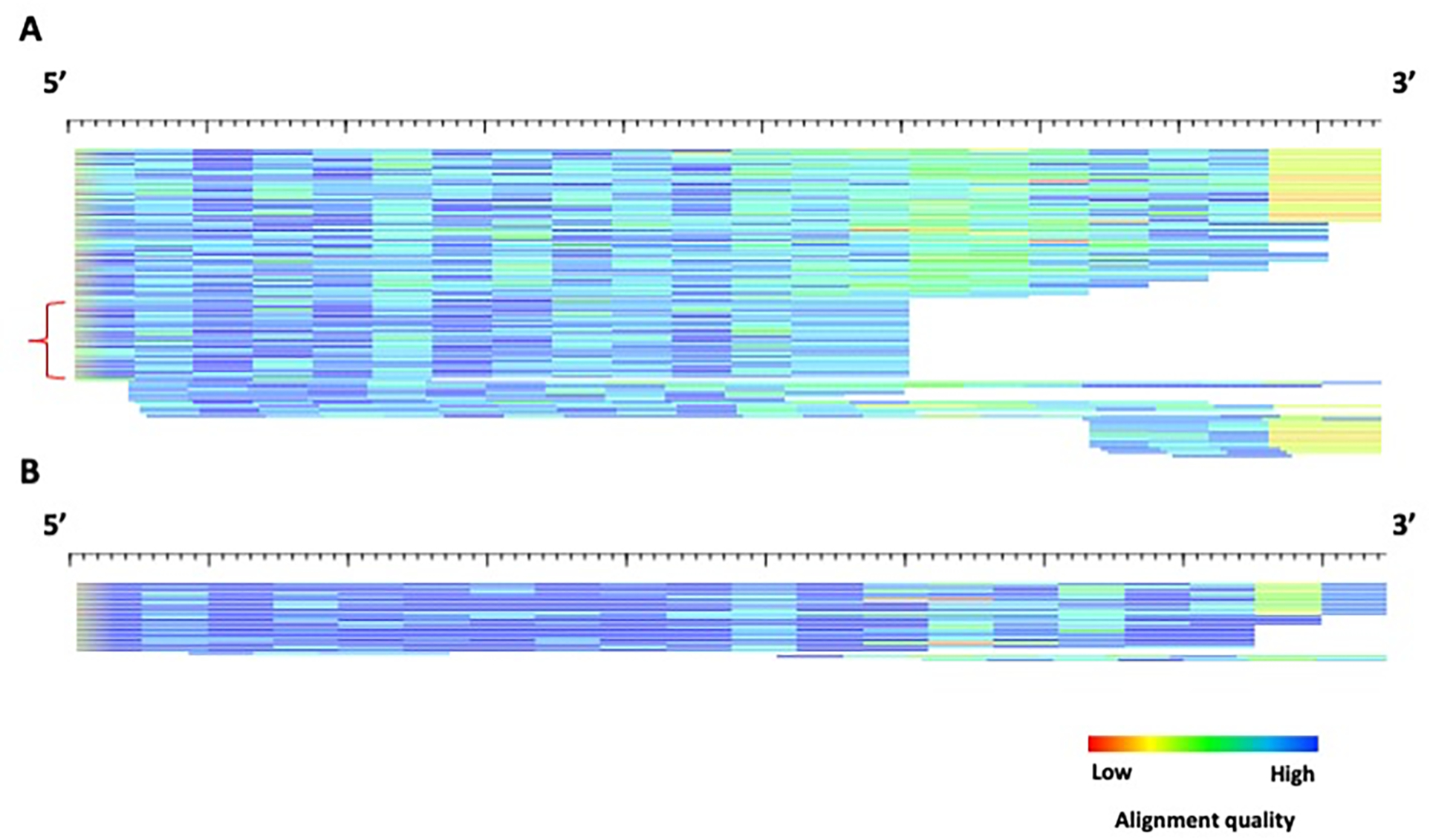
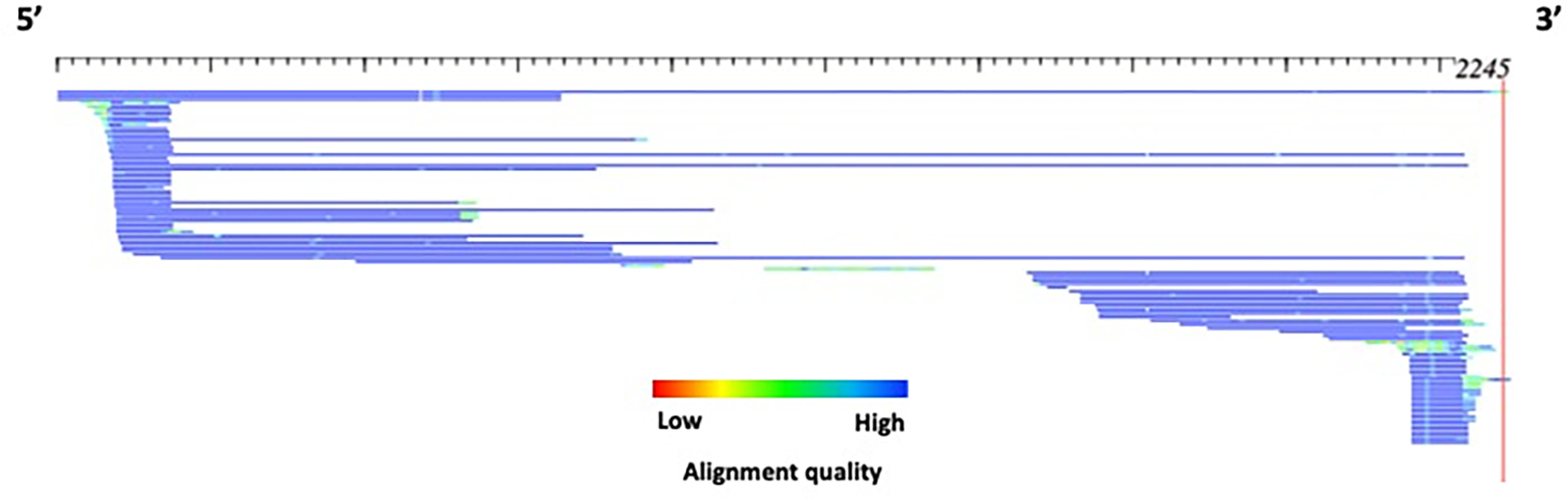
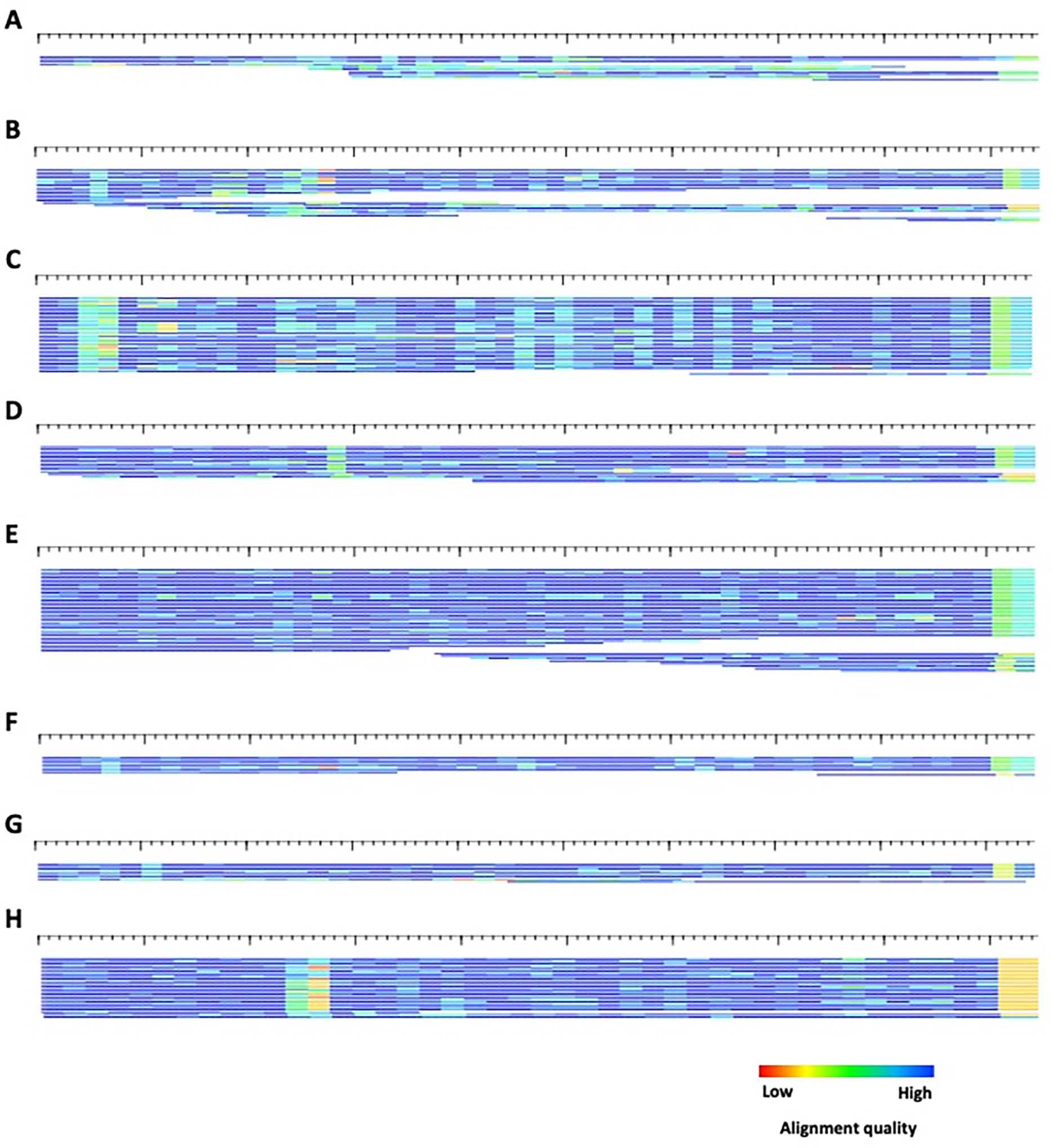
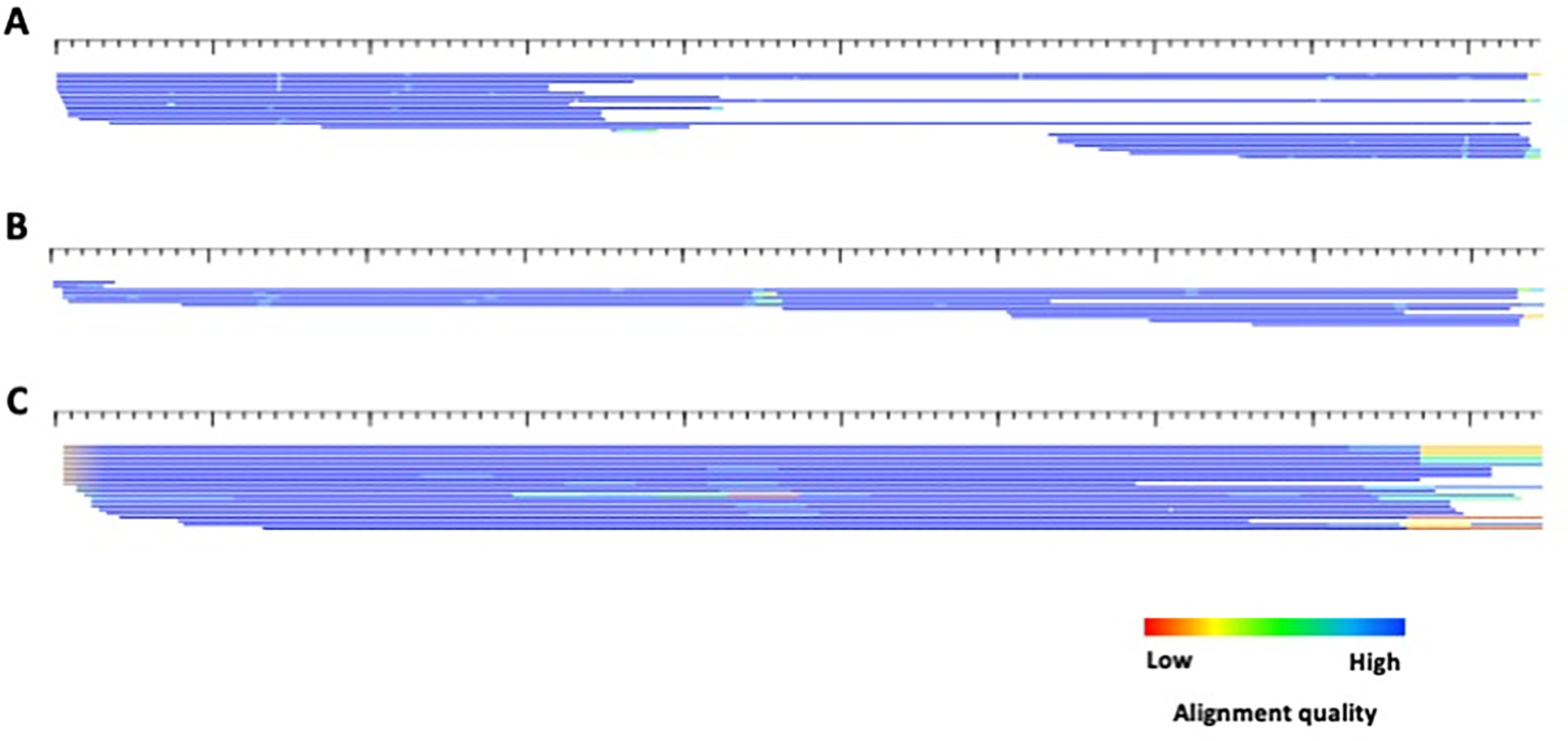
Transposable elements (TEs) have the ability to alter individual genomic landscapes and shape the course of evolution for species in which they reside. Such profound changes can be understood by studying the biology of the organism and the interplay of the TEs it hosts. Characterizing and curating TEs across a wide range of species is a fundamental first step in this endeavor. This protocol employs techniques honed while developing TE libraries for a wide range of organisms and specifically addresses: (1) the extension of truncated de novo results into full-length TE families; (2) the iterative refinement of TE multiple sequence alignments; and (3) the use of alignment visualization to assess model completeness and subfamily structure. © 2021 Wiley Periodicals LLC. Basic Protocol: Extension and edge polishing of consensi and seed alignments derived from de novo repeat finders Support Protocol: Generating seed alignments using a library of consensi and a genome assembly.
Keywords: RepeatMasker; RepeatModeler; alignment; curation; hidden Markov model; transposable elements.
© 2021 Wiley Periodicals LLC.
Figures






References
-
- Arensburger P, Piégu B, & Bigot Y (2016). The future of transposable element annotation and their classification in the light of functional genomics - what we can learn from the fables of Jean de la Fontaine? [Review of The future of transposable element annotation and their classification in the light of functional genomics - what we can learn from the fables of Jean de la Fontaine?]. Mobile Genetic Elements, 6(6), e1256852. - PMC - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
