

Learning the protein language: Evolution, structure, and function
- PMID: 34139171
- PMCID: PMC8238390
- DOI: 10.1016/j.cels.2021.05.017
Learning the protein language: Evolution, structure, and function
Abstract
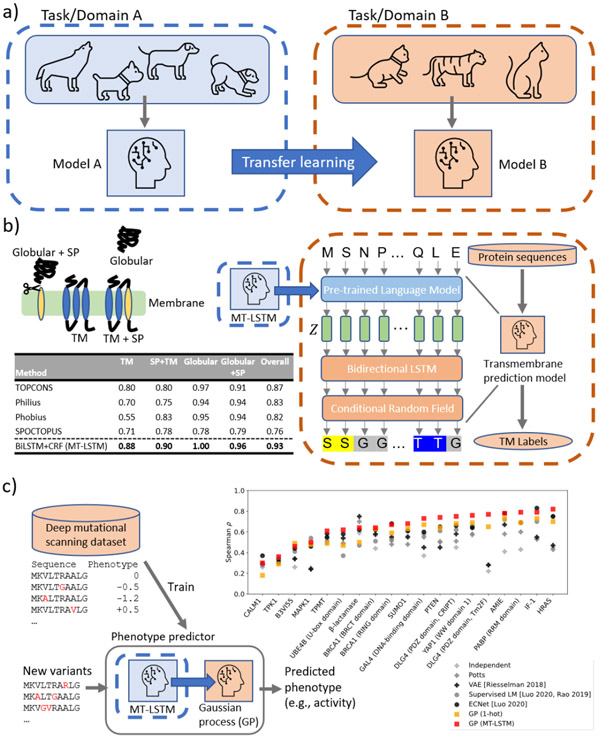
Language models have recently emerged as a powerful machine-learning approach for distilling information from massive protein sequence databases. From readily available sequence data alone, these models discover evolutionary, structural, and functional organization across protein space. Using language models, we can encode amino-acid sequences into distributed vector representations that capture their structural and functional properties, as well as evaluate the evolutionary fitness of sequence variants. We discuss recent advances in protein language modeling and their applications to downstream protein property prediction problems. We then consider how these models can be enriched with prior biological knowledge and introduce an approach for encoding protein structural knowledge into the learned representations. The knowledge distilled by these models allows us to improve downstream function prediction through transfer learning. Deep protein language models are revolutionizing protein biology. They suggest new ways to approach protein and therapeutic design. However, further developments are needed to encode strong biological priors into protein language models and to increase their accessibility to the broader community.
Keywords: contact prediction; deep neural networks; inductive bias; language models; natural language processing; protein sequences; proteins; transfer learning; transmembrane region prediction.
Copyright © 2021 Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of interests The authors declare no competing interests.
Figures
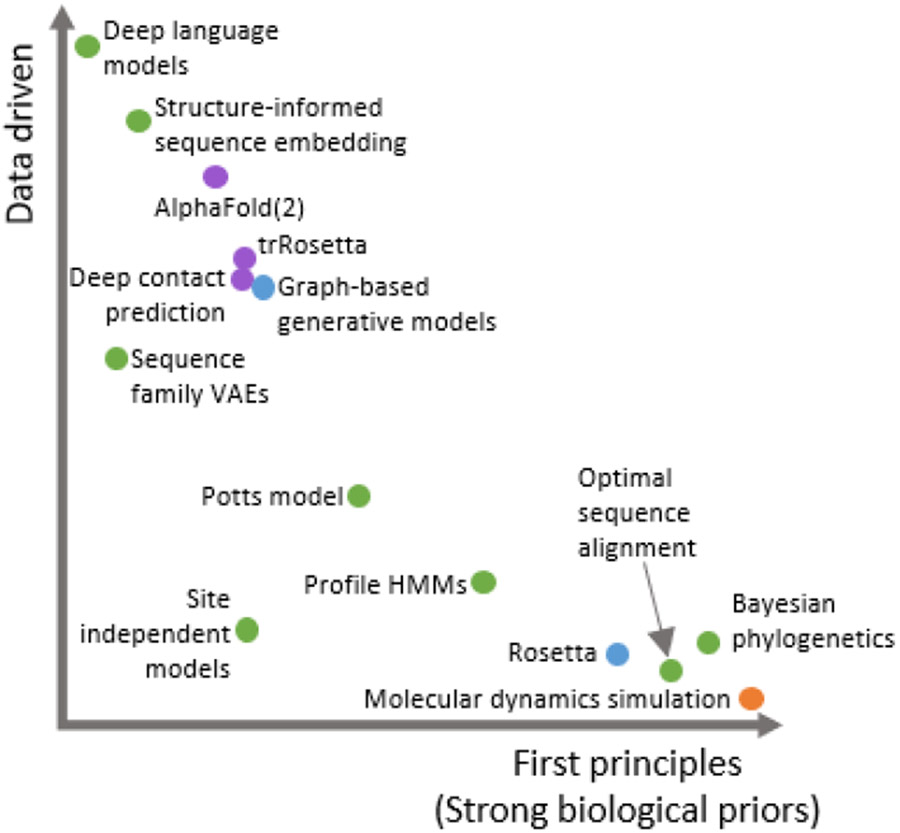
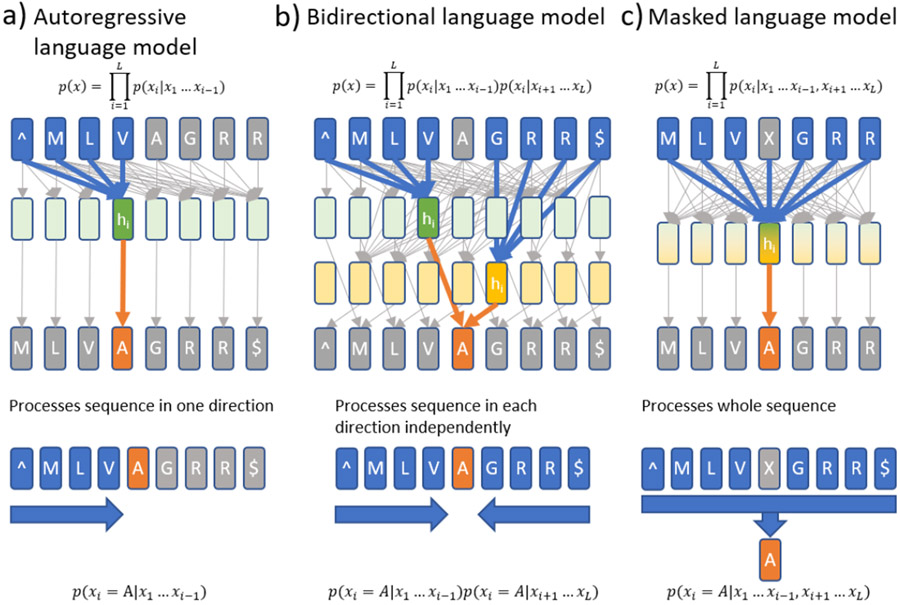
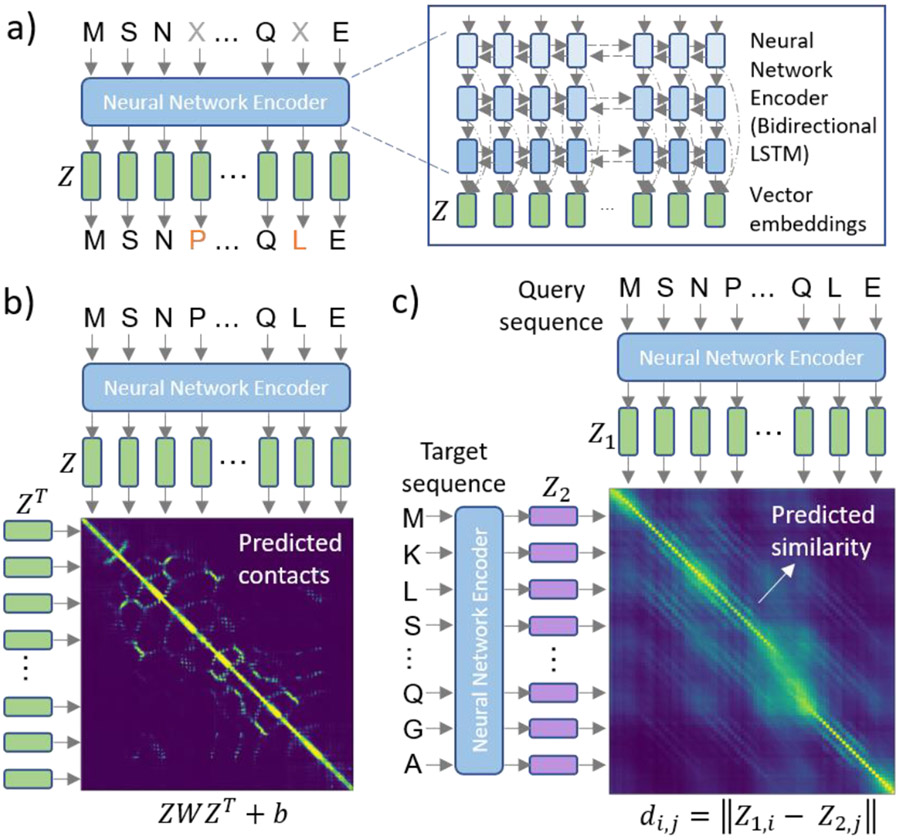
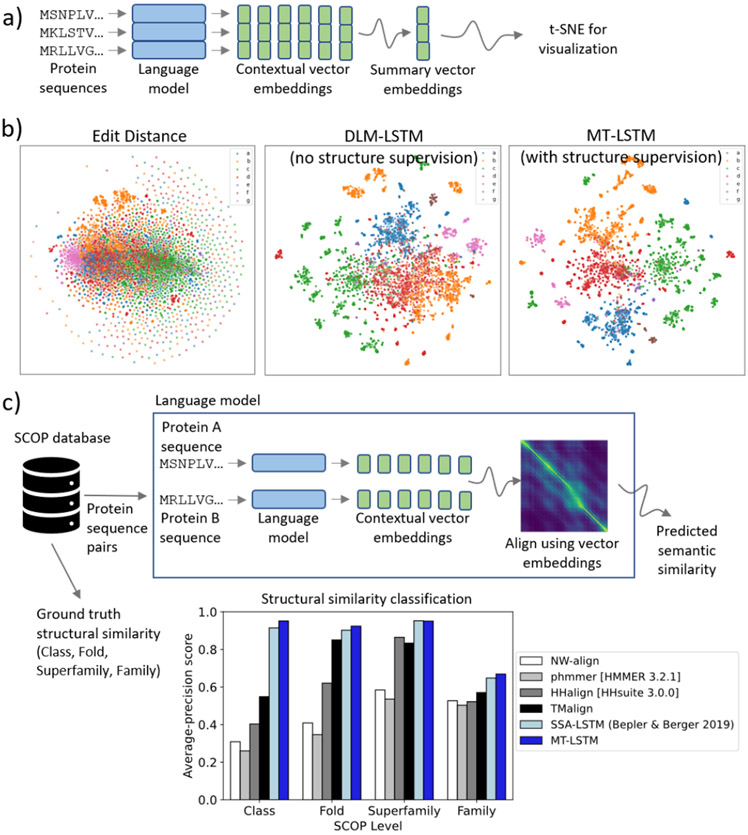
References
-
- Ekeberg M, Lövkvist C, Lan Y, Weigt M & Aurell E Improved contact prediction in proteins: using pseudolikelihoods to infer Potts models. Phys. Rev. E Stat. Nonlin. Soft Matter Phys 87, 012707 (2013). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
