

Williams syndrome
- PMID: 34140529
- PMCID: PMC9437774
- DOI: 10.1038/s41572-021-00276-z
Williams syndrome
Abstract
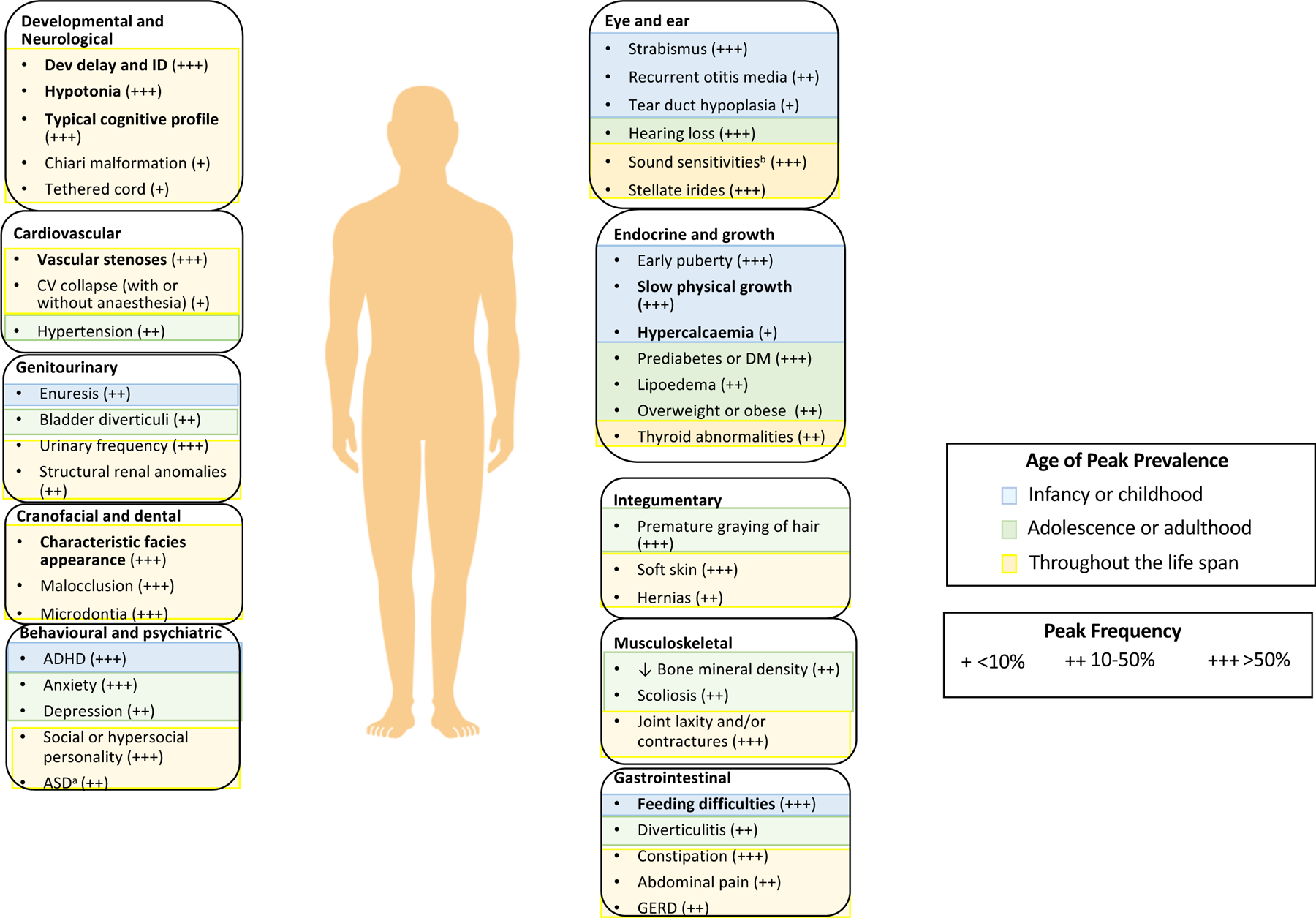
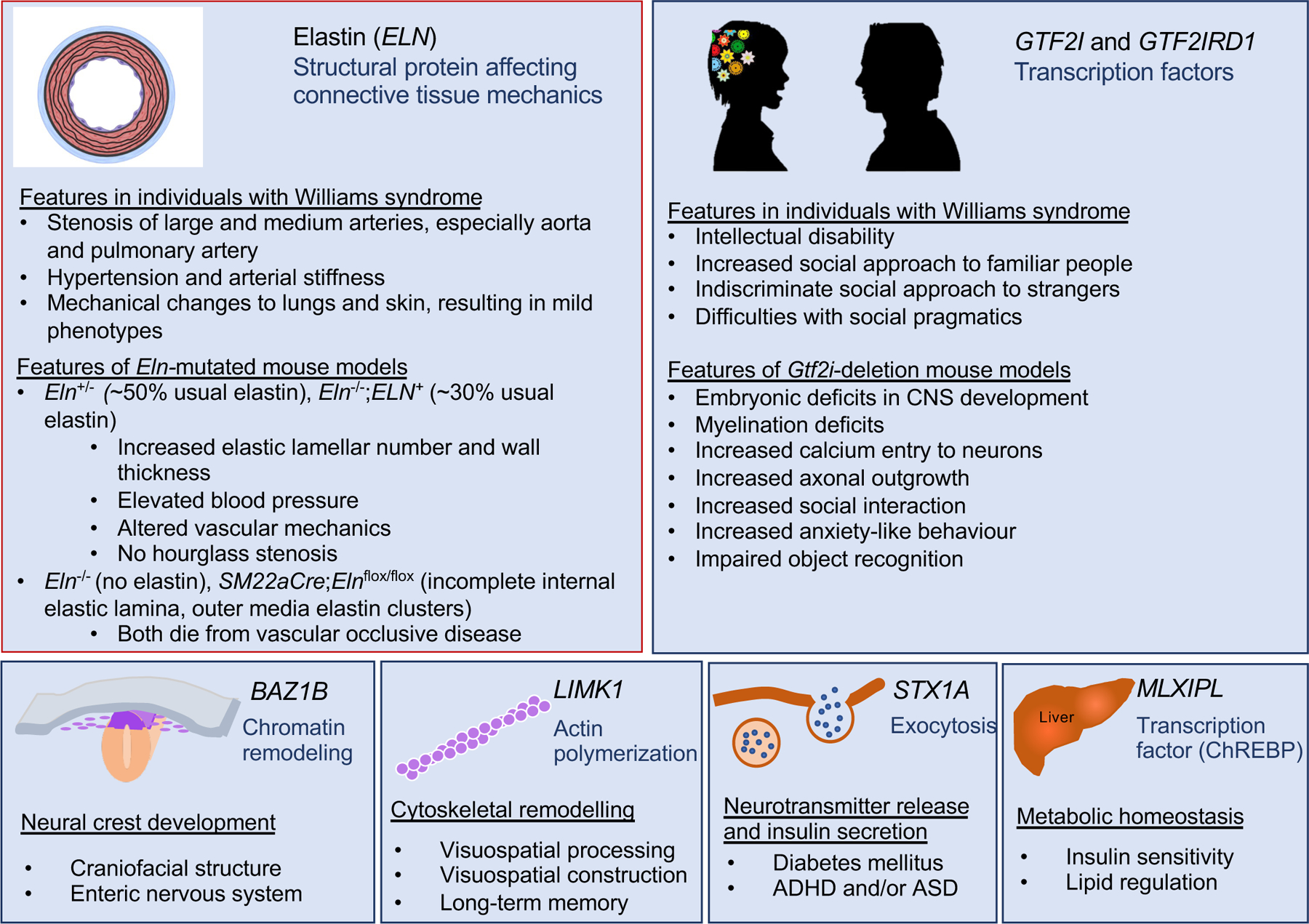
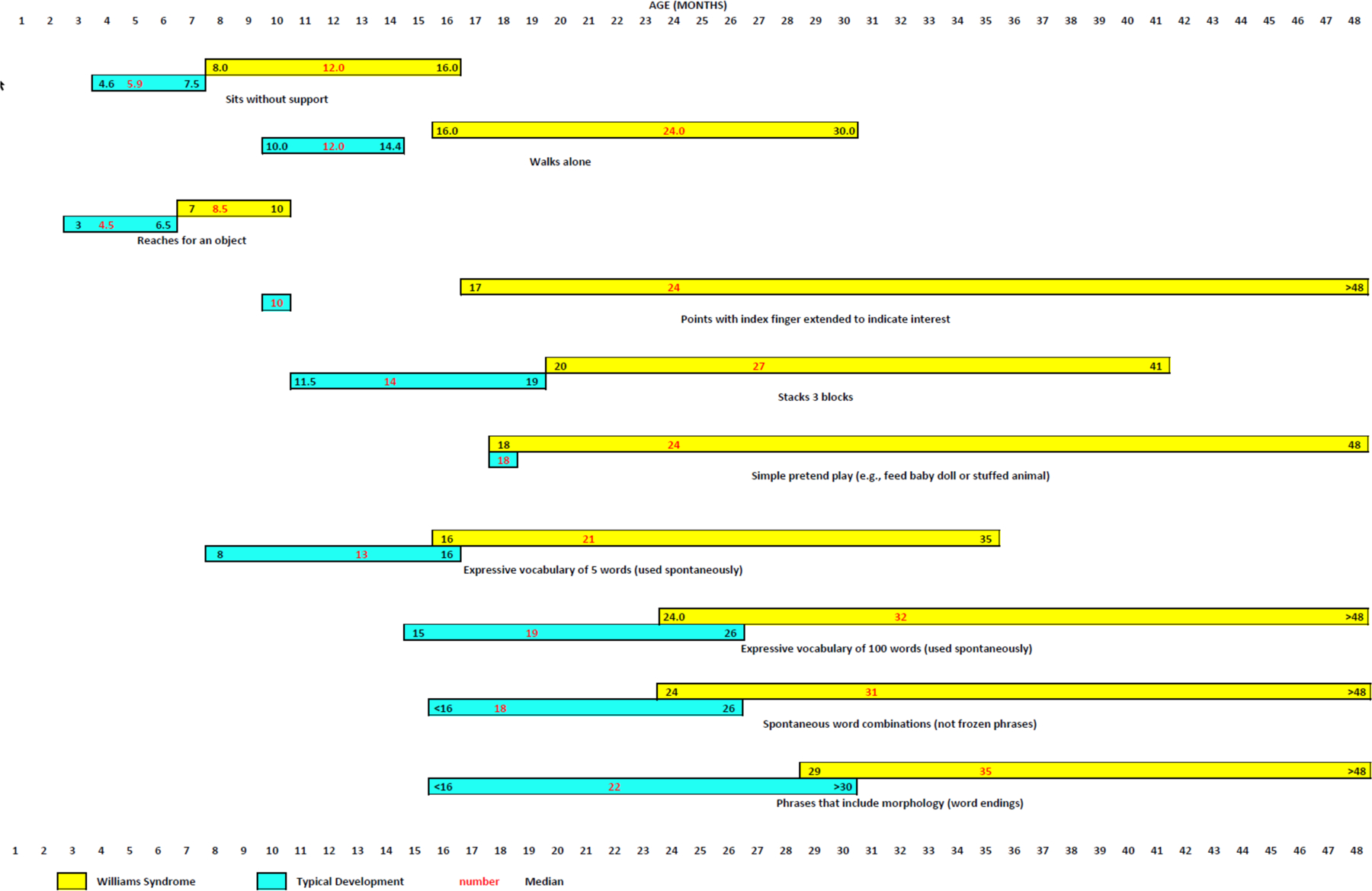
Williams syndrome (WS) is a relatively rare microdeletion disorder that occurs in as many as 1:7,500 individuals. WS arises due to the mispairing of low-copy DNA repetitive elements at meiosis. The deletion size is similar across most individuals with WS and leads to the loss of one copy of 25-27 genes on chromosome 7q11.23. The resulting unique disorder affects multiple systems, with cardinal features including but not limited to cardiovascular disease (characteristically stenosis of the great arteries and most notably supravalvar aortic stenosis), a distinctive craniofacial appearance, and a specific cognitive and behavioural profile that includes intellectual disability and hypersociability. Genotype-phenotype evidence is strongest for ELN, the gene encoding elastin, which is responsible for the vascular and connective tissue features of WS, and for the transcription factor genes GTF2I and GTF2IRD1, which are known to affect intellectual ability, social functioning and anxiety. Mounting evidence also ascribes phenotypic consequences to the deletion of BAZ1B, LIMK1, STX1A and MLXIPL, but more work is needed to understand the mechanism by which these deletions contribute to clinical outcomes. The age of diagnosis has fallen in regions of the world where technological advances, such as chromosomal microarray, enable clinicians to make the diagnosis of WS without formally suspecting it, allowing earlier intervention by medical and developmental specialists. Phenotypic variability is considerable for all cardinal features of WS but the specific sources of this variability remain unknown. Further investigation to identify the factors responsible for these differences may lead to mechanism-based rather than symptom-based therapies and should therefore be a high research priority.
Conflict of interest statement
Competing interests
The authors declare no conflicts of interest.
Figures

References
-
- Fanconi G, Girardet P, Schlesinger B, Butler N & Black J Chronische Hypercalcaemie kombiniert mit Osteosklerose, Hyperazotaemie, Minderwuchs, und kongenitalen Missbildungen [Chronic hypercalcemia, combined with osteosclerosis, hyperazotemia, nanism, and congenital malformations]. Helvetica Paediatrica Acta, 314–349 (1952). - PubMed
-
-
Del Pasqua A et al. New findings concerning cardiovascular manifestations emerging from long-term follow-up of 150 patients with the Williams-Beuren-Beuren syndrome. Cardiol Young 19, 563–567, doi: 10.1017/S1047951109990837 (2009).
This paper reports the cardiovascular outcomes from a large number of people with WS over a range of 0.5–25 years (average 6 years).
-
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
