

Molecular mechanisms underlying nucleotide repeat expansion disorders
- PMID: 34140671
- PMCID: PMC9612635
- DOI: 10.1038/s41580-021-00382-6
Molecular mechanisms underlying nucleotide repeat expansion disorders
Erratum in
-
Author Correction: Molecular mechanisms underlying nucleotide repeat expansion disorders.Nat Rev Mol Cell Biol. 2021 Sep;22(9):644. doi: 10.1038/s41580-021-00396-0. Nat Rev Mol Cell Biol. 2021. PMID: 34230651 No abstract available.
Abstract
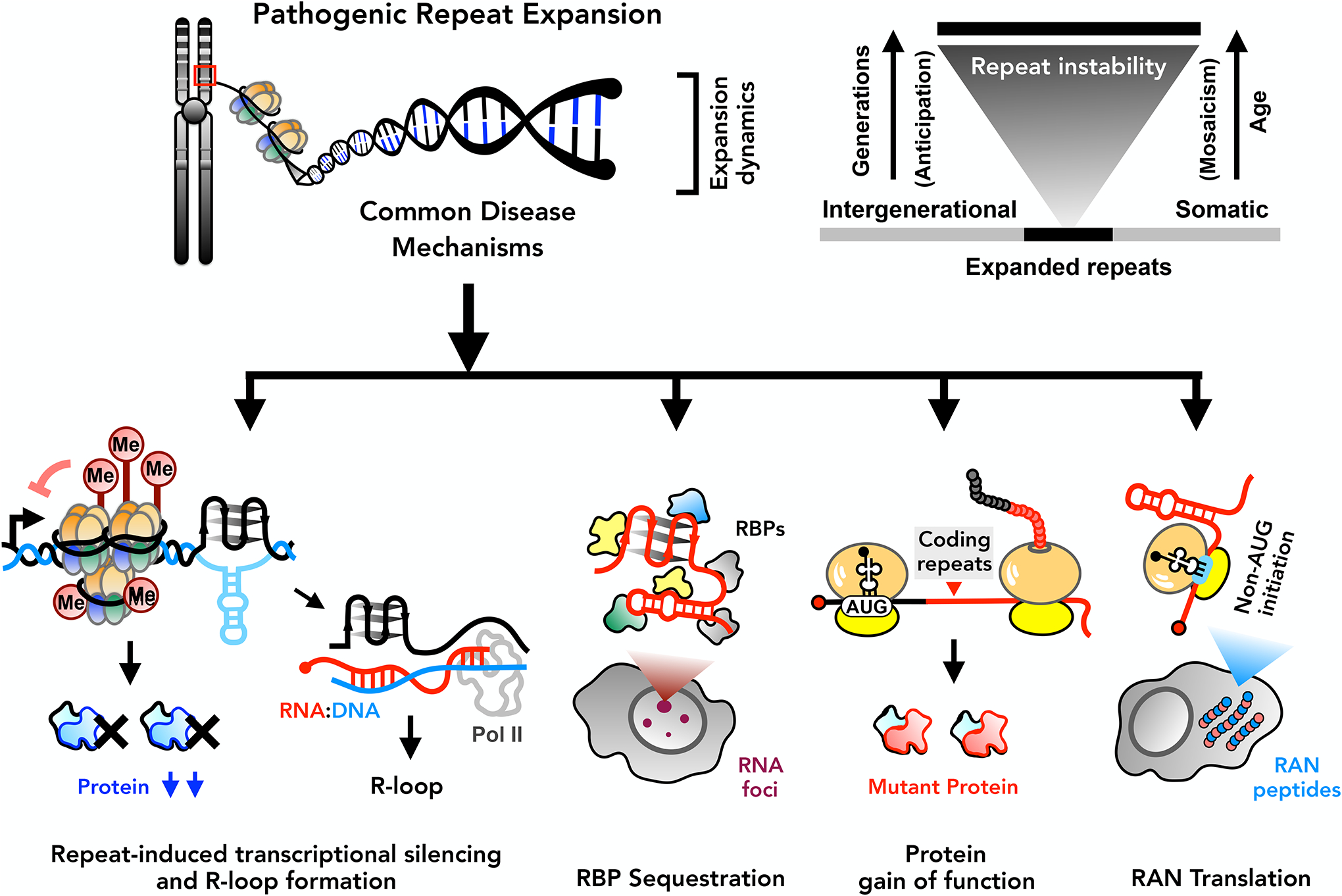
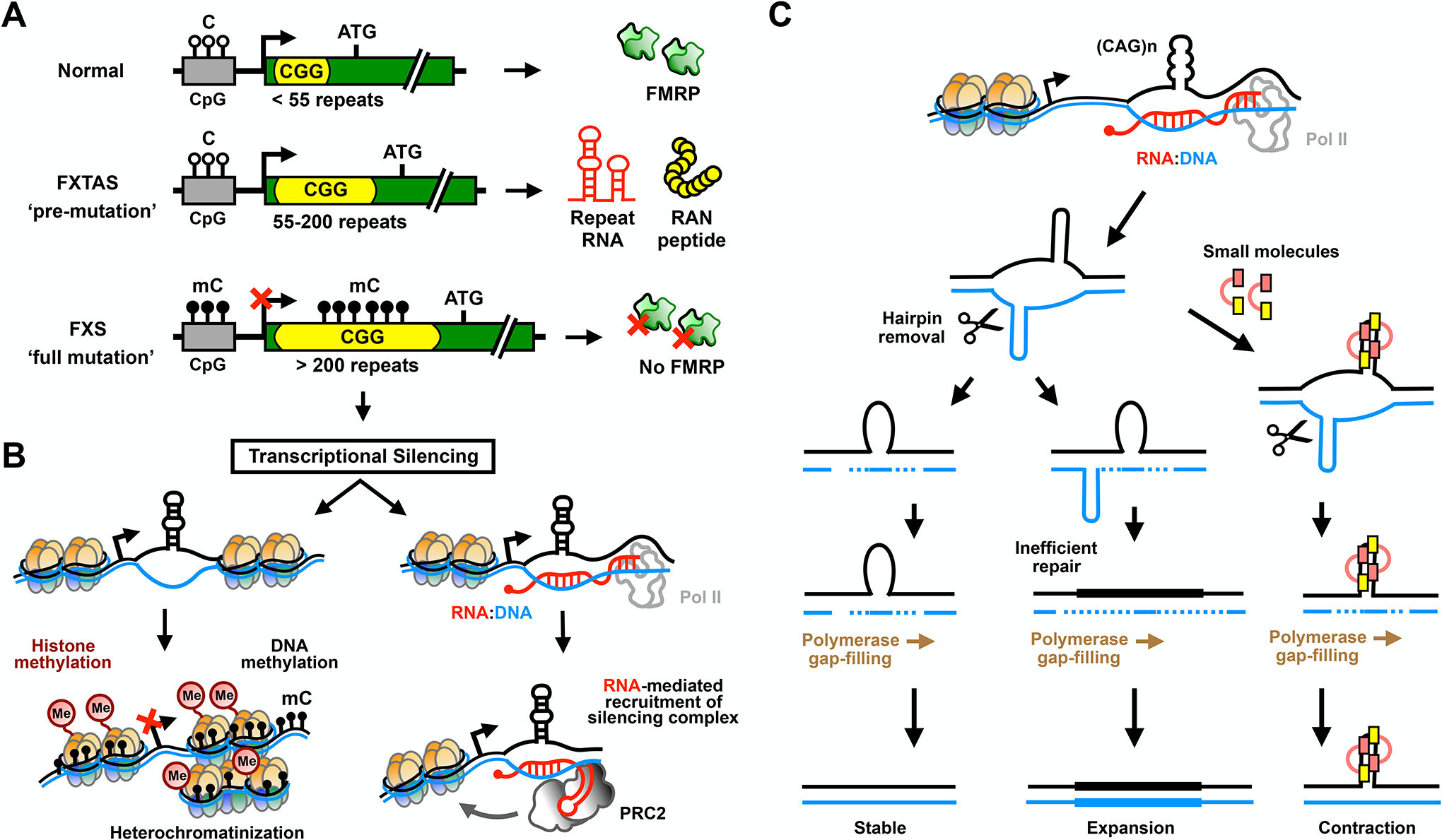
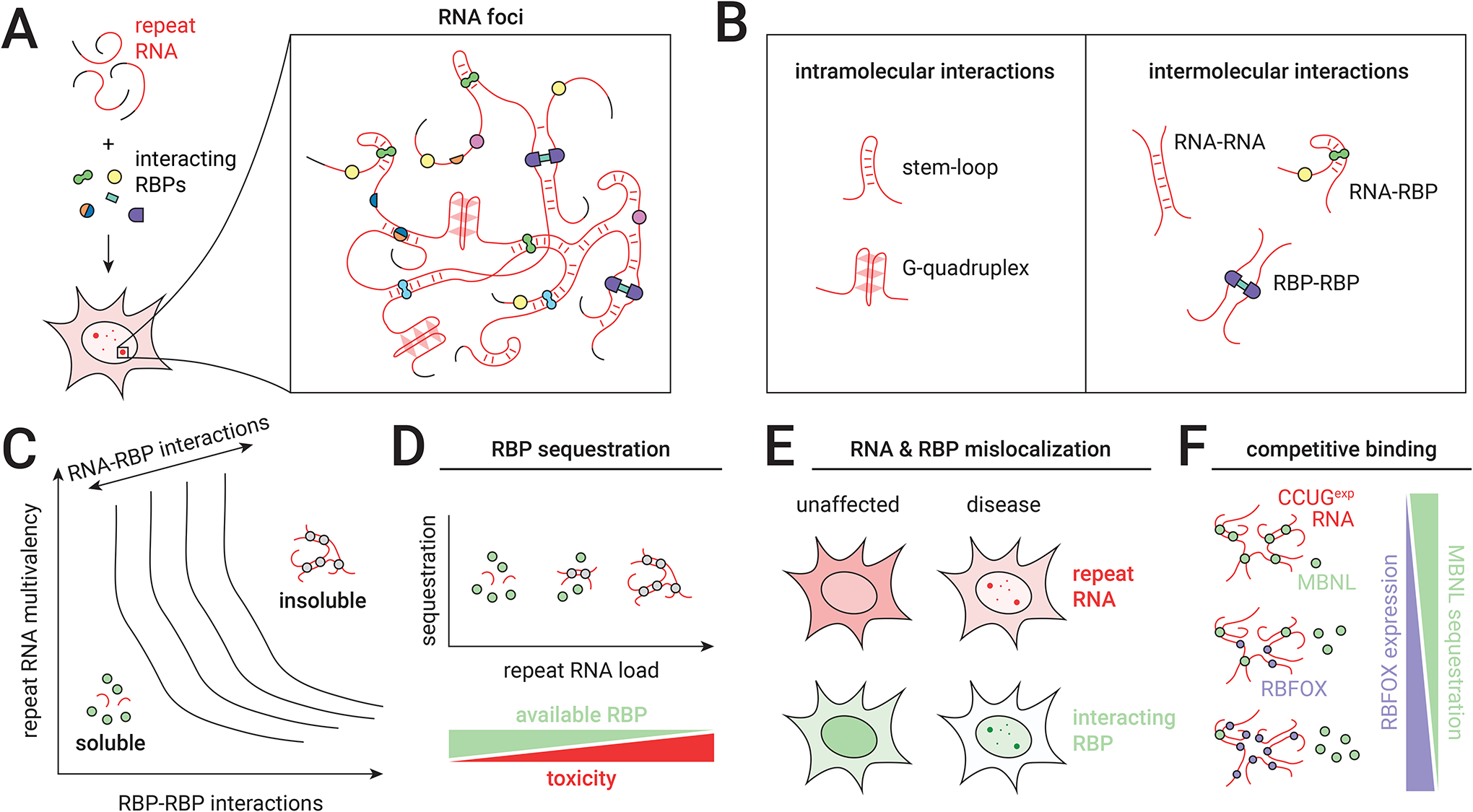
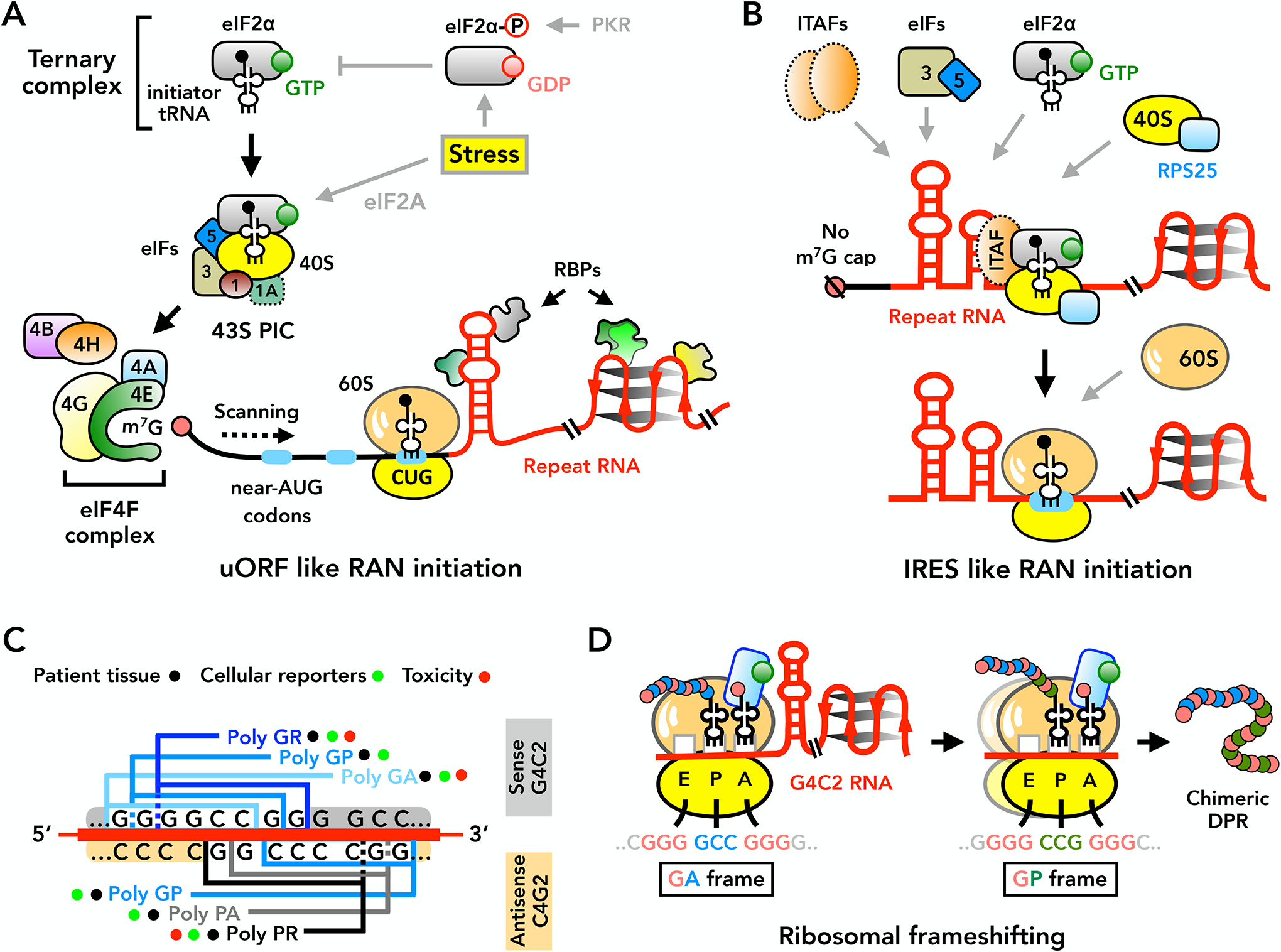
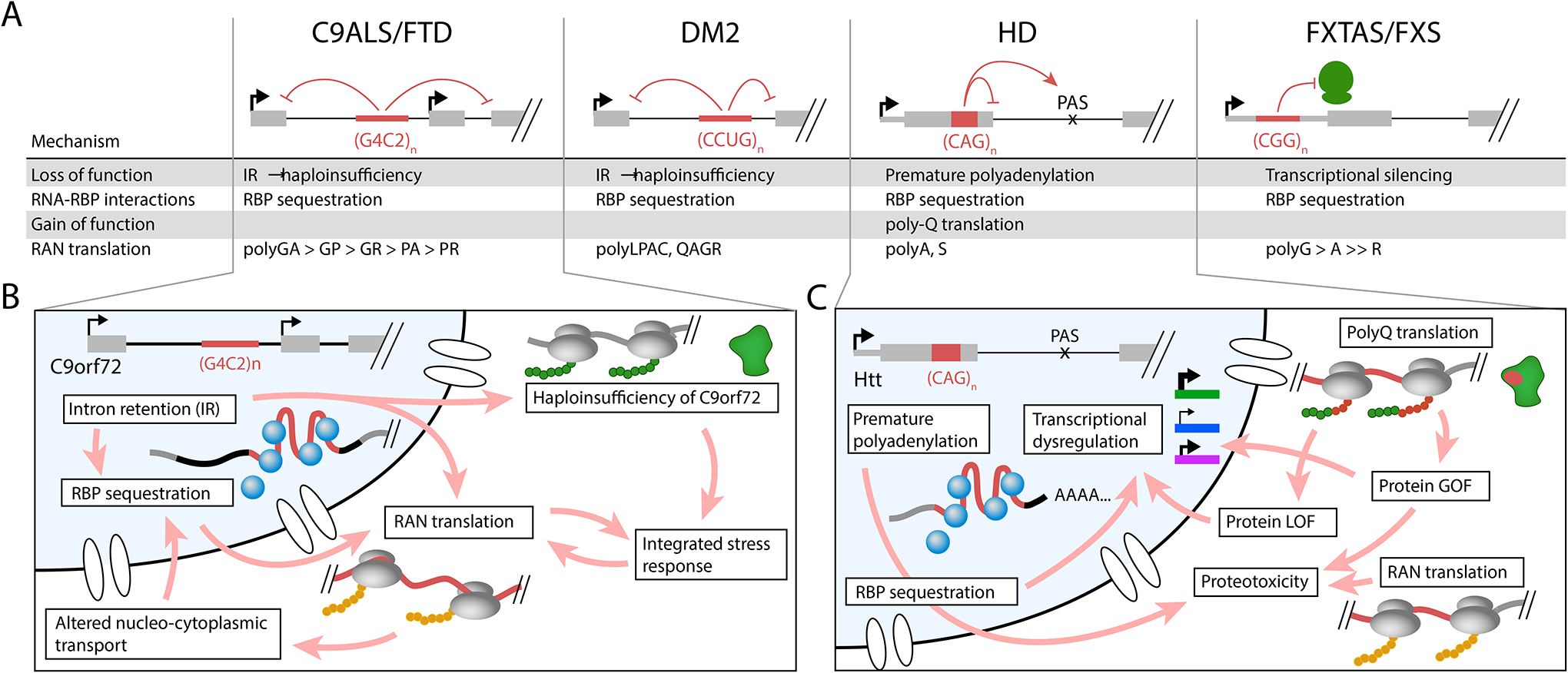
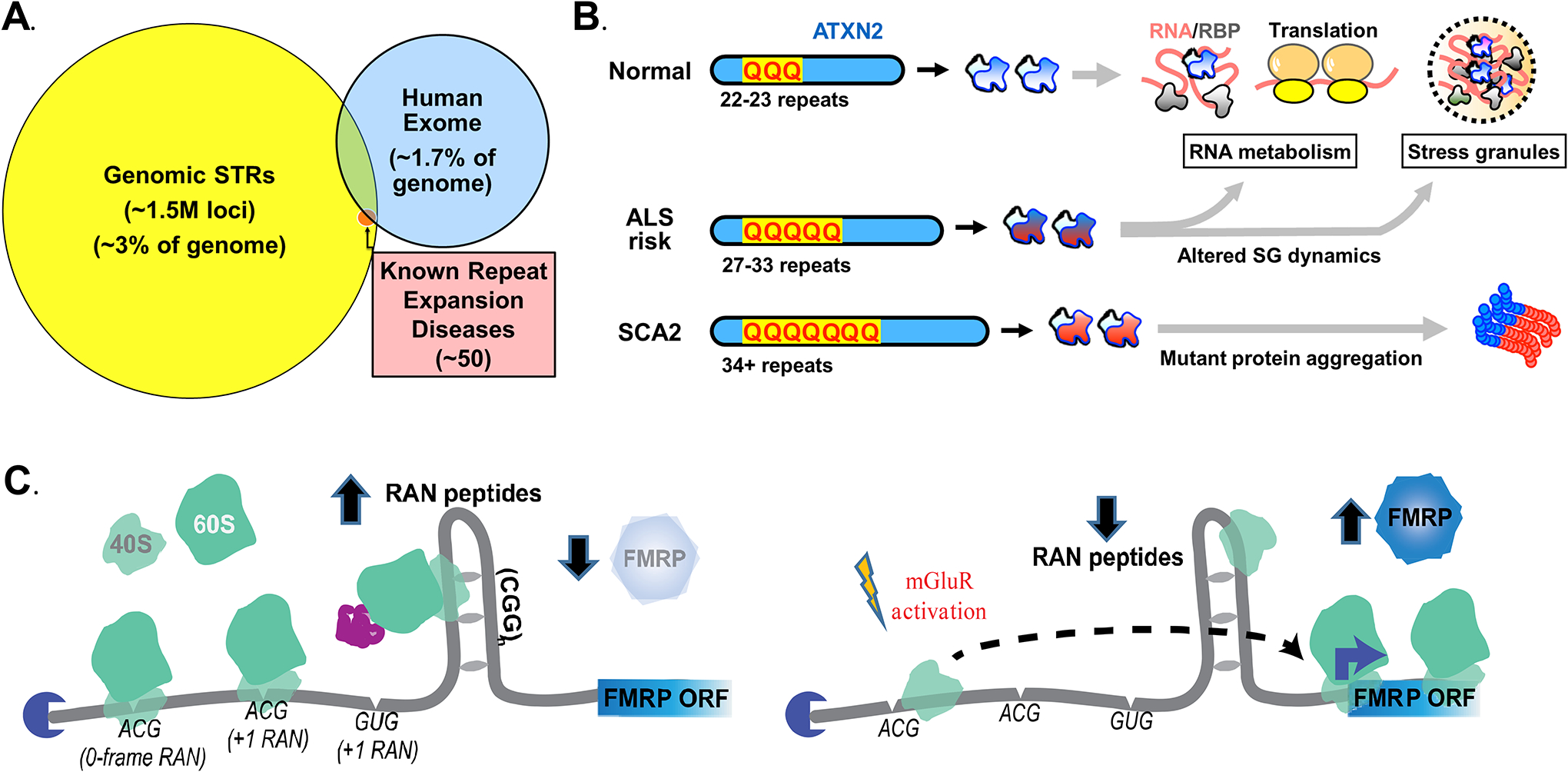
The human genome contains over one million short tandem repeats. Expansion of a subset of these repeat tracts underlies over fifty human disorders, including common genetic causes of amyotrophic lateral sclerosis (ALS) and frontotemporal dementia (C9orf72), polyglutamine-associated ataxias and Huntington disease, myotonic dystrophy, and intellectual disability disorders such as Fragile X syndrome. In this Review, we discuss the four major mechanisms by which expansion of short tandem repeats causes disease: loss of function through transcription repression, RNA-mediated gain of function through gelation and sequestration of RNA-binding proteins, gain of function of canonically translated repeat-harbouring proteins, and repeat-associated non-AUG translation of toxic repeat peptides. Somatic repeat instability amplifies these mechanisms and influences both disease age of onset and tissue specificity of pathogenic features. We focus on the crosstalk between these disease mechanisms, and argue that they often synergize to drive pathogenesis. We also discuss the emerging native functions of repeat elements and how their dynamics might contribute to disease at a larger scale than currently appreciated. Lastly, we propose that lynchpins tying these disease mechanisms and native functions together offer promising therapeutic targets with potential shared applications across this class of human disorders.
© 2021. Springer Nature Limited.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
