

Integrating single-cell and spatial transcriptomics to elucidate intercellular tissue dynamics
- PMID: 34145435
- PMCID: PMC9888017
- DOI: 10.1038/s41576-021-00370-8
Integrating single-cell and spatial transcriptomics to elucidate intercellular tissue dynamics
Abstract
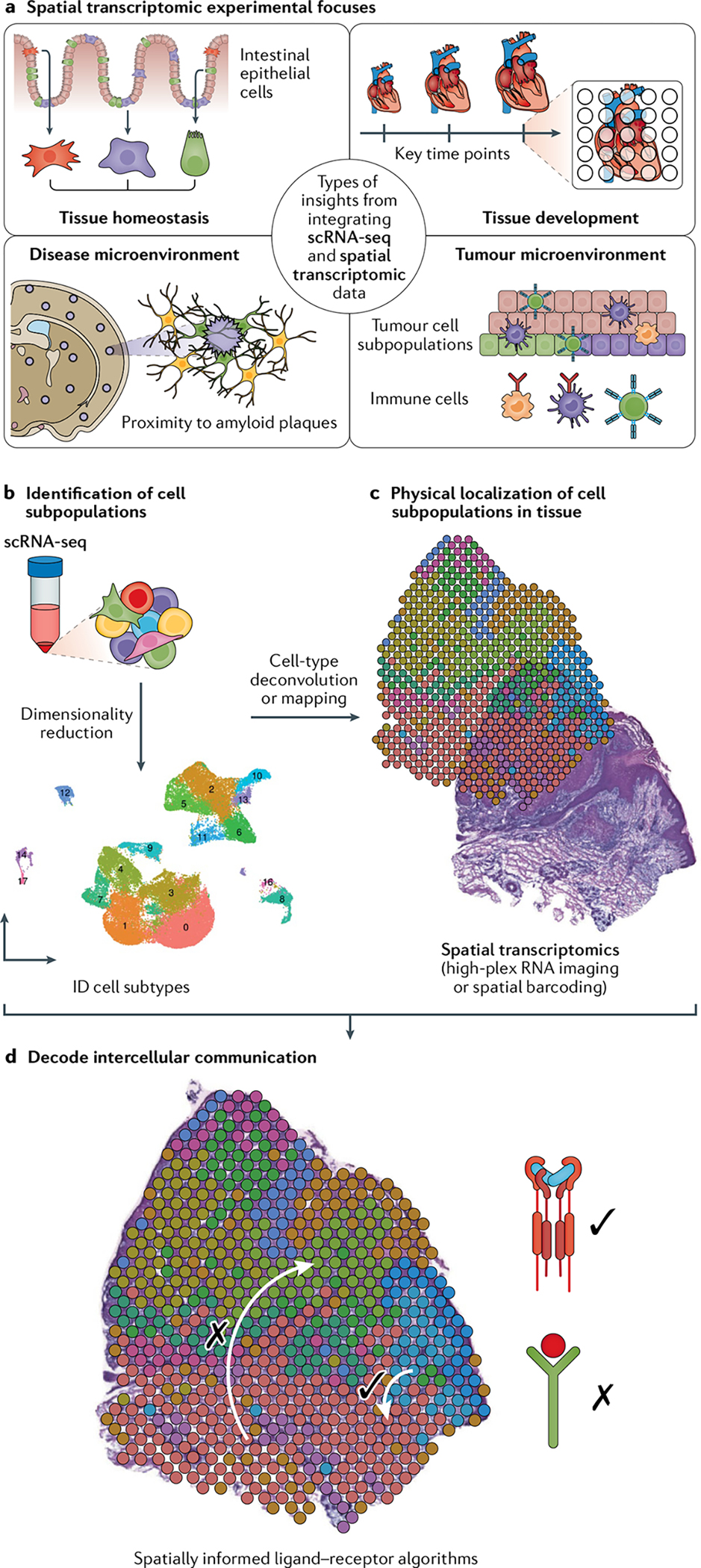
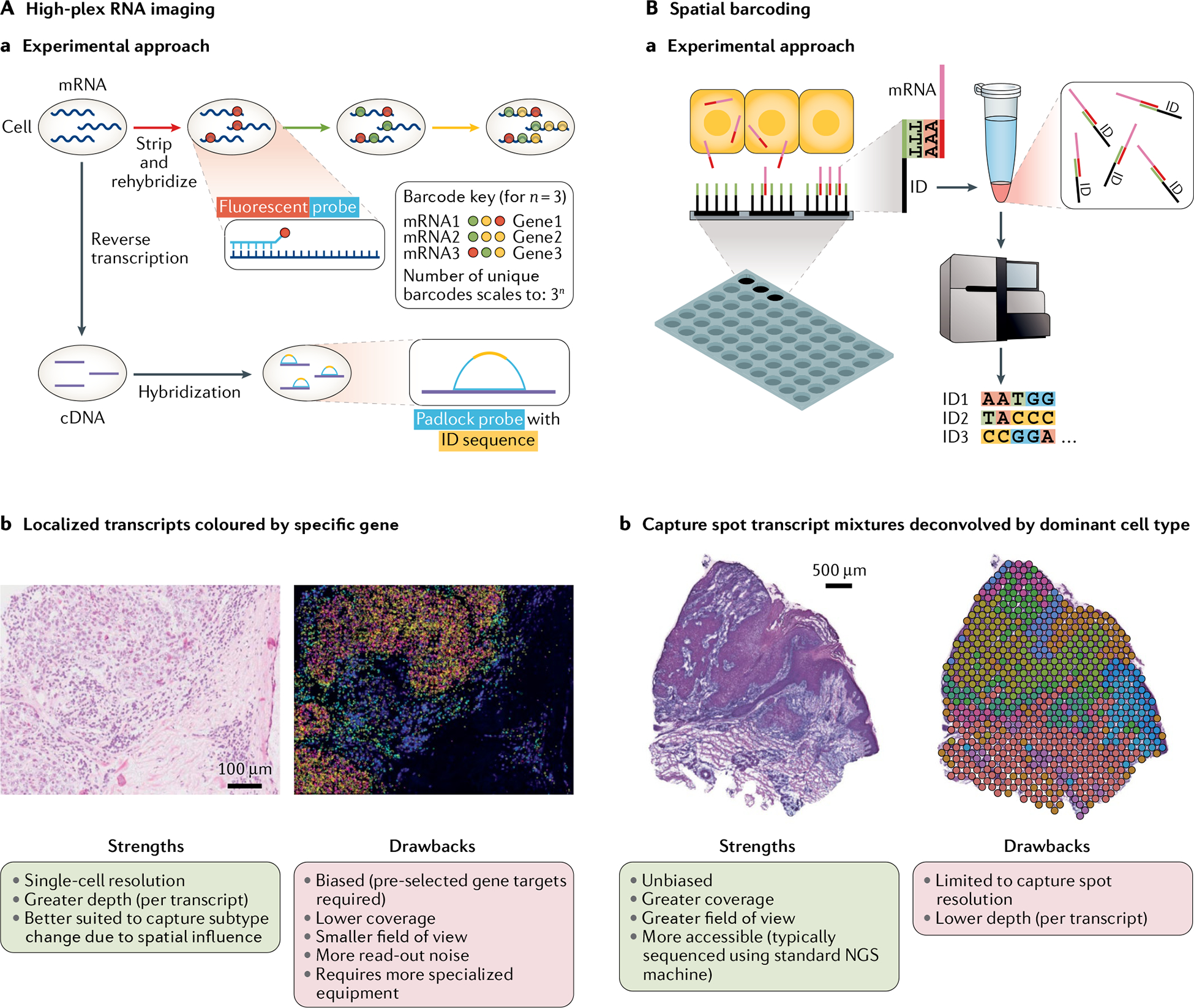
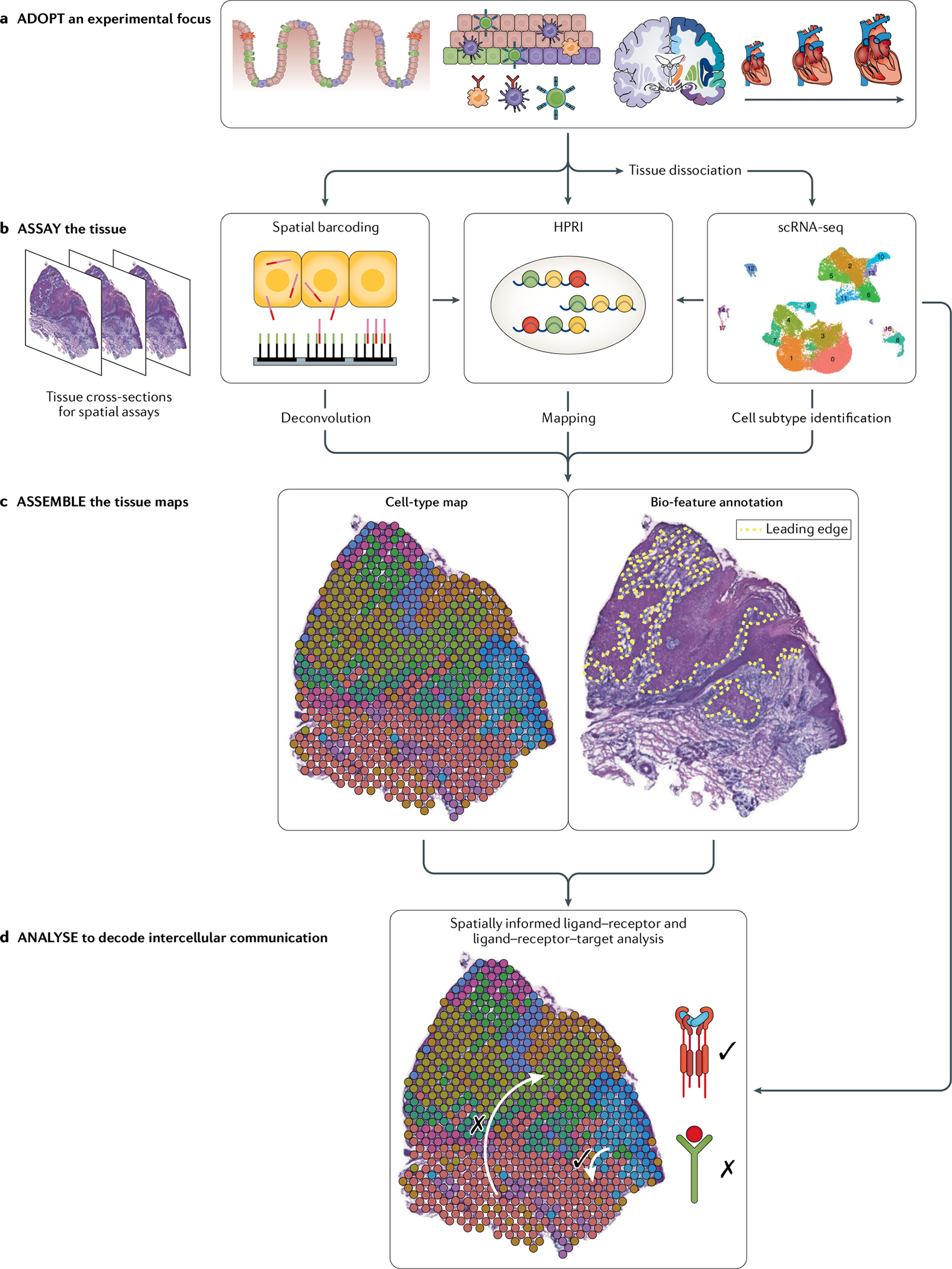
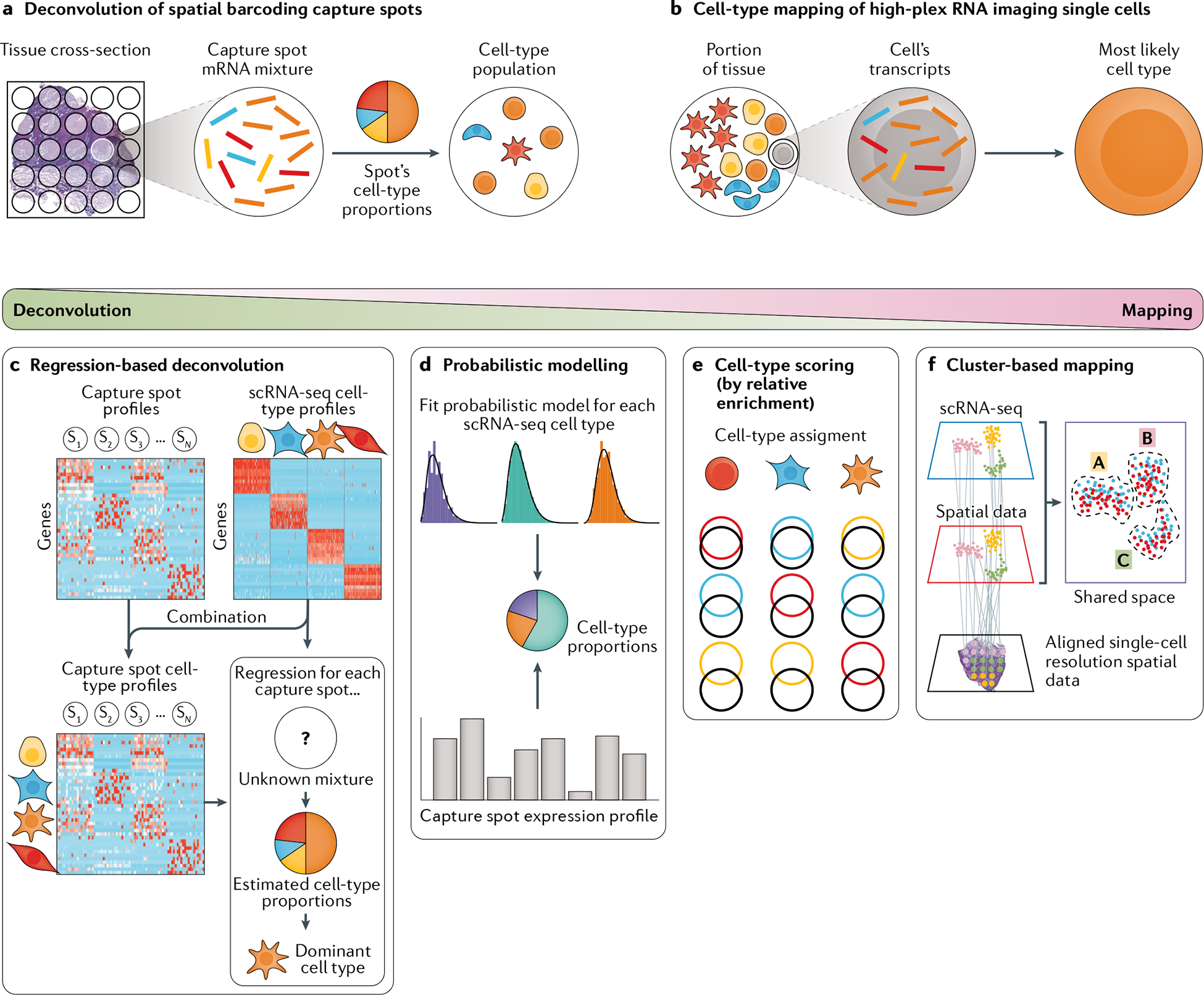
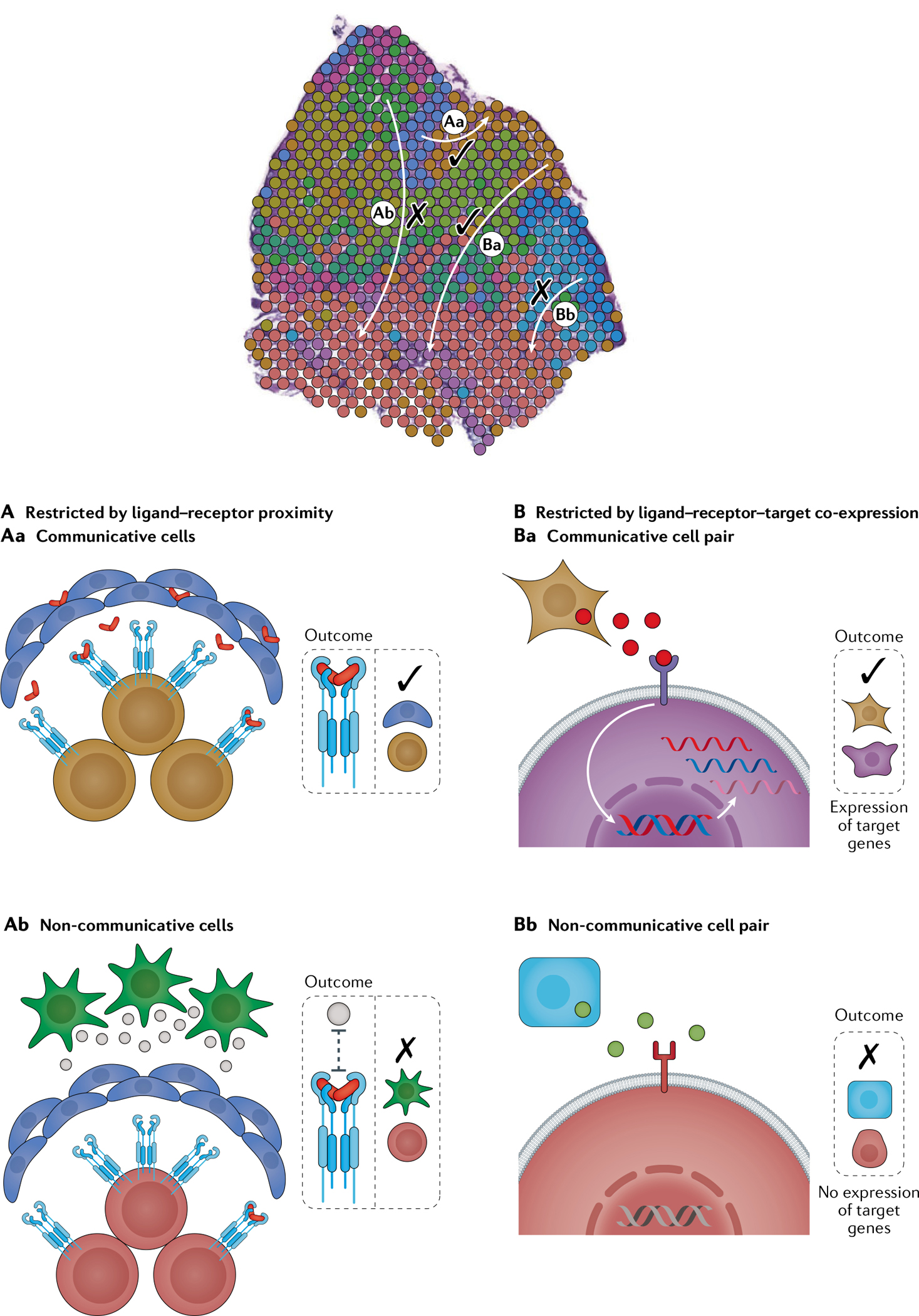
Single-cell RNA sequencing (scRNA-seq) identifies cell subpopulations within tissue but does not capture their spatial distribution nor reveal local networks of intercellular communication acting in situ. A suite of recently developed techniques that localize RNA within tissue, including multiplexed in situ hybridization and in situ sequencing (here defined as high-plex RNA imaging) and spatial barcoding, can help address this issue. However, no method currently provides as complete a scope of the transcriptome as does scRNA-seq, underscoring the need for approaches to integrate single-cell and spatial data. Here, we review efforts to integrate scRNA-seq with spatial transcriptomics, including emerging integrative computational methods, and propose ways to effectively combine current methodologies.
© 2021. This is a U.S. government work and not under copyright protection in the U.S.; foreign copyright protection may apply.
Conflict of interest statement
Competing interests
The authors declare no competing interests.
Figures
References
-
- [No authors listed] Method of the Year 2020: spatially resolved transcriptomics. Nat. Methods 18, 1 (2021). - PubMed
-
- Stegle O, Teichmann S & Marioni J Computational and analytical challenges in single-cell transcriptomics. Nat. Rev. Genet. 16, 133–145 (2015). - PubMed
-
- Tang F et al. mRNA-seq whole-transcriptome analysis of a single cell. Nat. Methods 6, 377–382 (2009). - PubMed
-
- van den Brink SC et al. Single-cell sequencing reveals dissociation-induced gene expression in tissue subpopulations. Nat. Methods 14, 935–936 (2017). - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
