

Personalized Management of Pheochromocytoma and Paraganglioma
- PMID: 34147030
- PMCID: PMC8905338
- DOI: 10.1210/endrev/bnab019
Personalized Management of Pheochromocytoma and Paraganglioma
Erratum in
-
Erratum to: Personalized Management of Pheochromocytoma and Paraganglioma.Endocr Rev. 2022 Mar 9;43(2):440. doi: 10.1210/endrev/bnab044. Endocr Rev. 2022. PMID: 34904637 Free PMC article. No abstract available.
-
Corrigendum to: Personalized Management of Pheochromocytoma and Paraganglioma.Endocr Rev. 2022 Mar 9;43(2):437-439. doi: 10.1210/endrev/bnab045. Endocr Rev. 2022. PMID: 34904662 Free PMC article. No abstract available.
Abstract
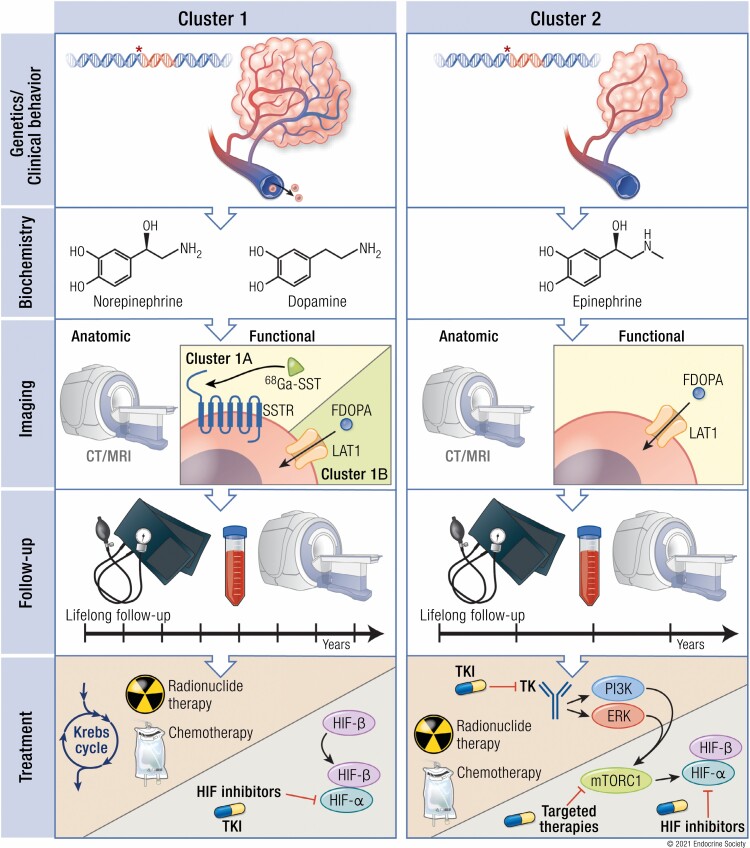
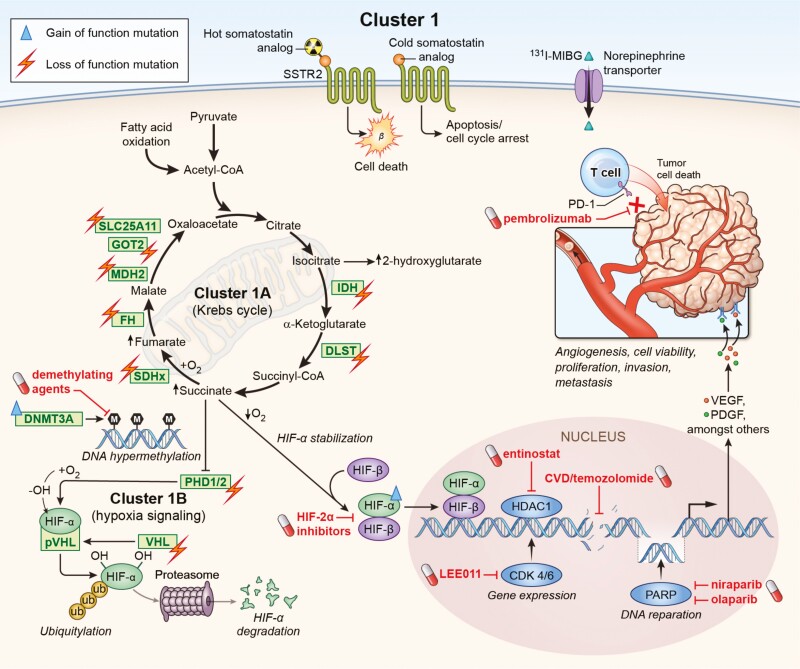
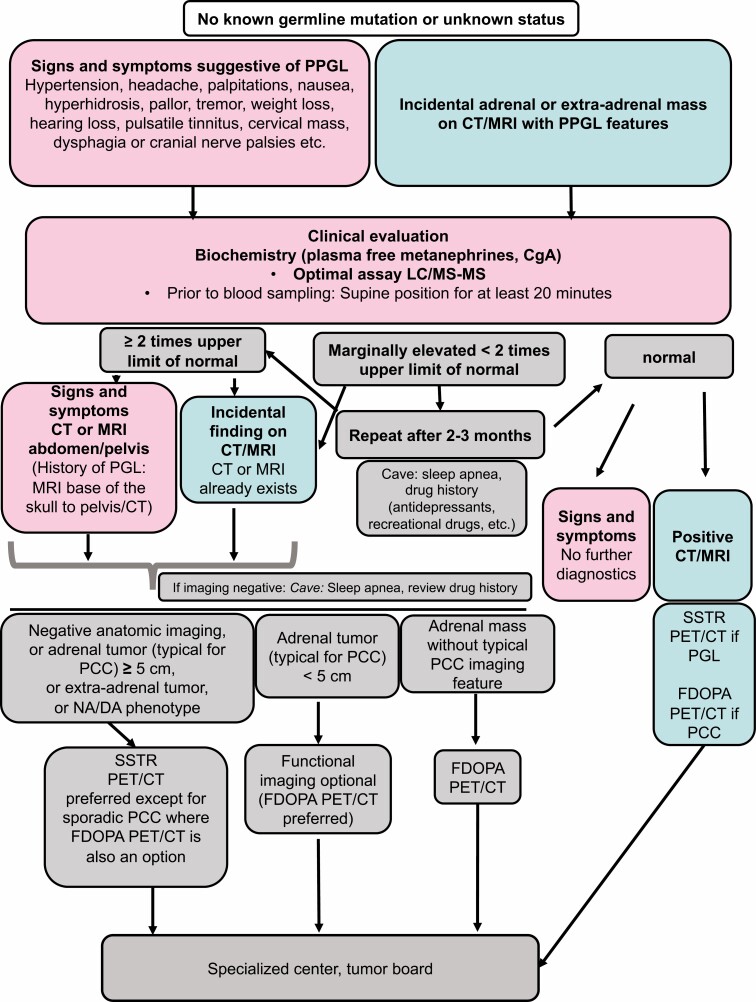
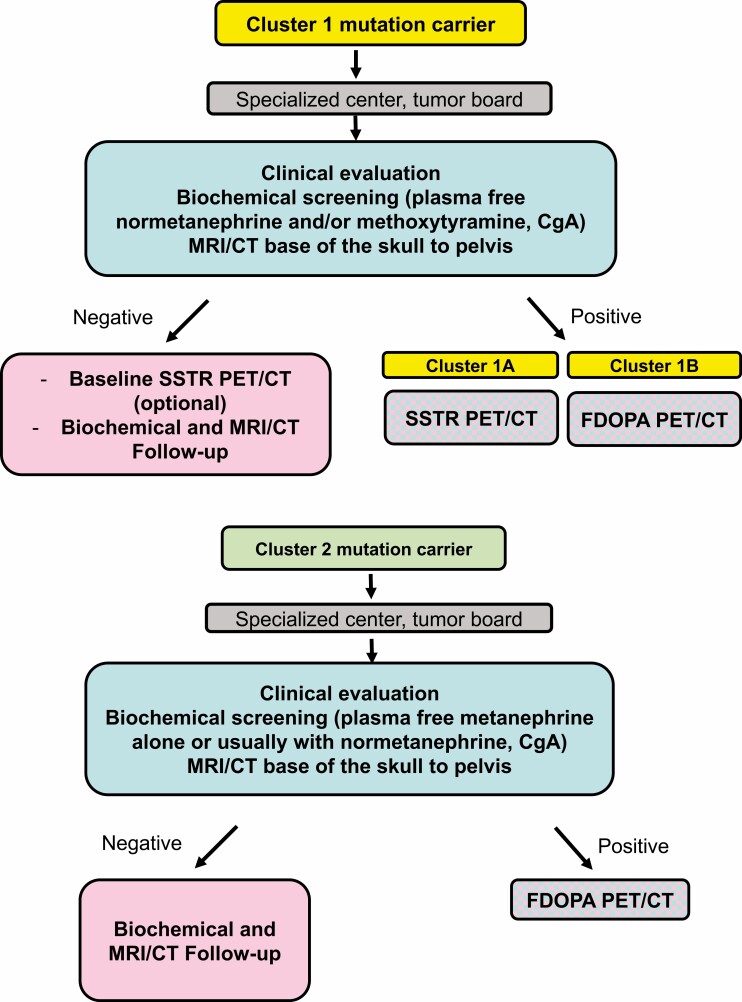
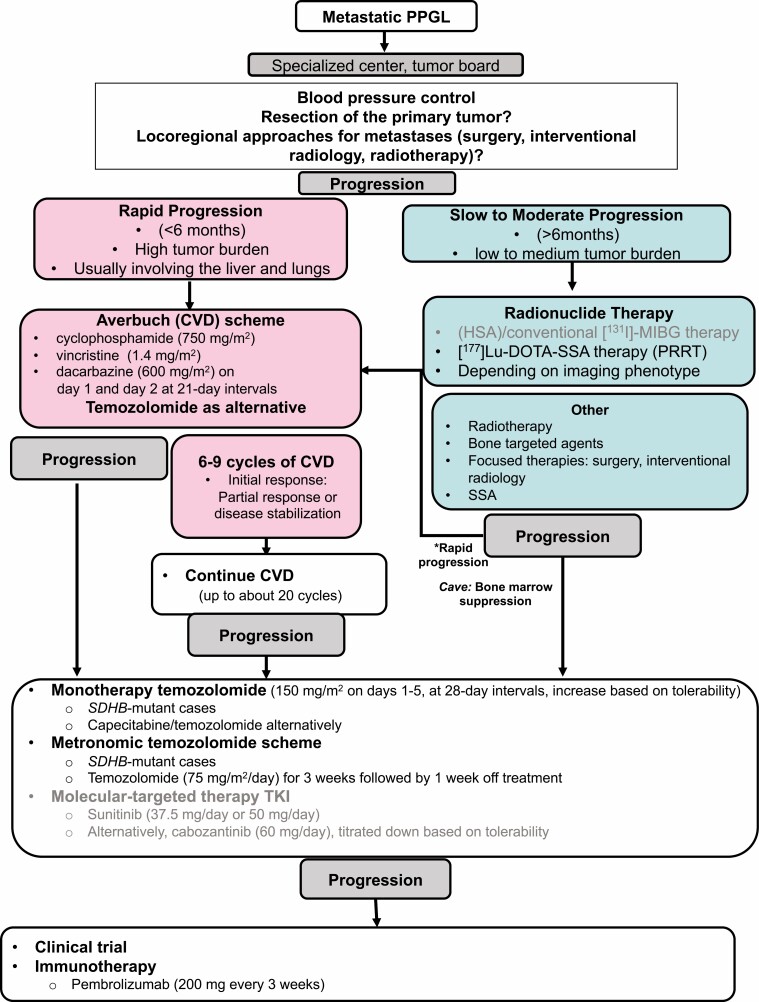
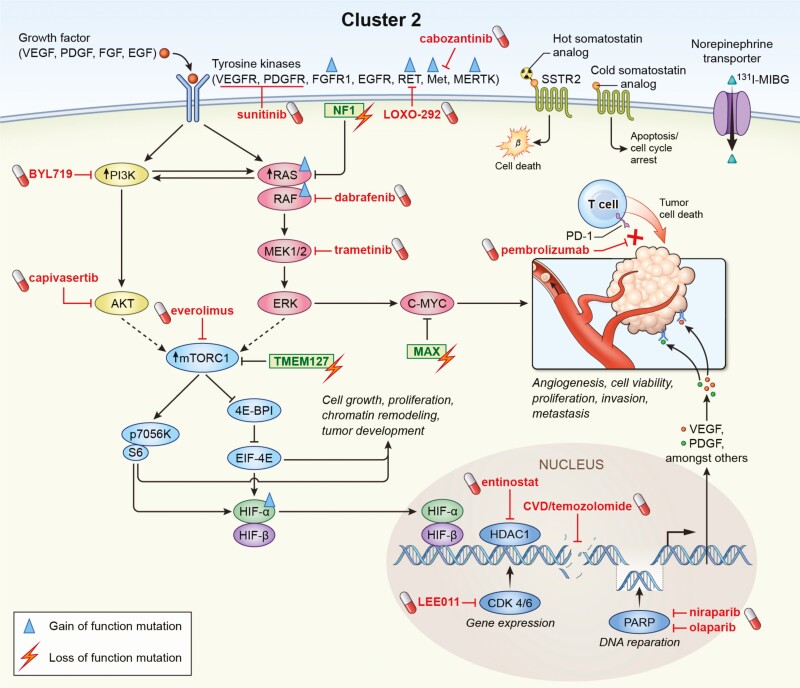
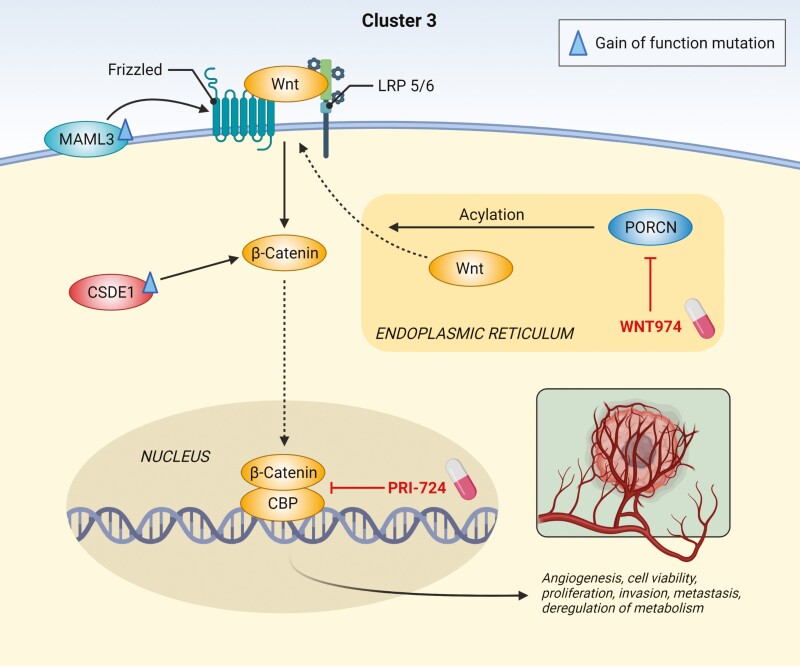
Pheochromocytomas/paragangliomas are characterized by a unique molecular landscape that allows their assignment to clusters based on underlying genetic alterations. With around 30% to 35% of Caucasian patients (a lower percentage in the Chinese population) showing germline mutations in susceptibility genes, pheochromocytomas/paragangliomas have the highest rate of heritability among all tumors. A further 35% to 40% of Caucasian patients (a higher percentage in the Chinese population) are affected by somatic driver mutations. Thus, around 70% of all patients with pheochromocytoma/paraganglioma can be assigned to 1 of 3 main molecular clusters with different phenotypes and clinical behavior. Krebs cycle/VHL/EPAS1-related cluster 1 tumors tend to a noradrenergic biochemical phenotype and require very close follow-up due to the risk of metastasis and recurrence. In contrast, kinase signaling-related cluster 2 tumors are characterized by an adrenergic phenotype and episodic symptoms, with generally a less aggressive course. The clinical correlates of patients with Wnt signaling-related cluster 3 tumors are currently poorly described, but aggressive behavior seems likely. In this review, we explore and explain why cluster-specific (personalized) management of pheochromocytoma/paraganglioma is essential to ascertain clinical behavior and prognosis, guide individual diagnostic procedures (biochemical interpretation, choice of the most sensitive imaging modalities), and provide personalized management and follow-up. Although cluster-specific therapy of inoperable/metastatic disease has not yet entered routine clinical practice, we suggest that informed personalized genetic-driven treatment should be implemented as a logical next step. This review amalgamates published guidelines and expert views within each cluster for a coherent individualized patient management plan.
Keywords: diagnostics; follow-up; molecular cluster; paraganglioma; pheochromocytoma; treatment.
© The Author(s) 2021. Published by Oxford University Press on behalf of the Endocrine Society.
Figures
References
-
- Lenders JWM, Kerstens MN, Amar L, et al. . Genetics, diagnosis, management and future directions of research of phaeochromocytoma and paraganglioma: a position statement and consensus of the Working Group on Endocrine Hypertension of the European Society of Hypertension. J Hypertens. 2020;38(8):1443-1456. - PMC - PubMed
-
- Lam AK. Update on adrenal tumours in 2017 World Health Organization (WHO) of endocrine tumours. Endocr Pathol. 2017;28(3):213-227. - PubMed
-
- Schovanek J, Martucci V, Wesley R, et al. . The size of the primary tumor and age at initial diagnosis are independent predictors of the metastatic behavior and survival of patients with SDHB-related pheochromocytoma and paraganglioma: a retrospective cohort study. BMC Cancer. 2014;14:523. - PMC - PubMed
