

Oligogenic Inheritance of Monoallelic TRIP11, FKBP10, NEK1, TBX5, and NBAS Variants Leading to a Phenotype Similar to Odontochondrodysplasia
- PMID: 34149817
- PMCID: PMC8206634
- DOI: 10.3389/fgene.2021.680838
Oligogenic Inheritance of Monoallelic TRIP11, FKBP10, NEK1, TBX5, and NBAS Variants Leading to a Phenotype Similar to Odontochondrodysplasia
Abstract
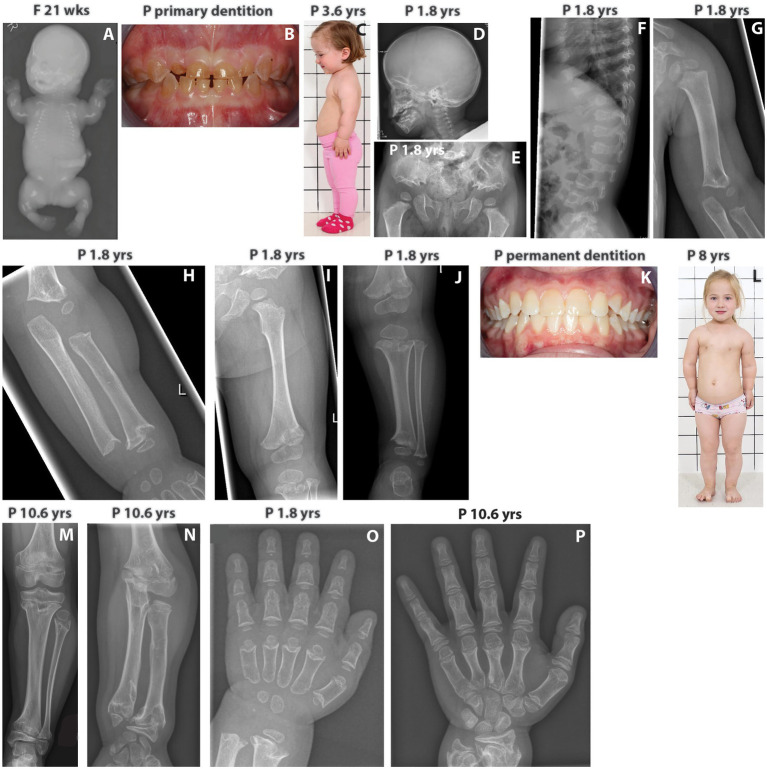
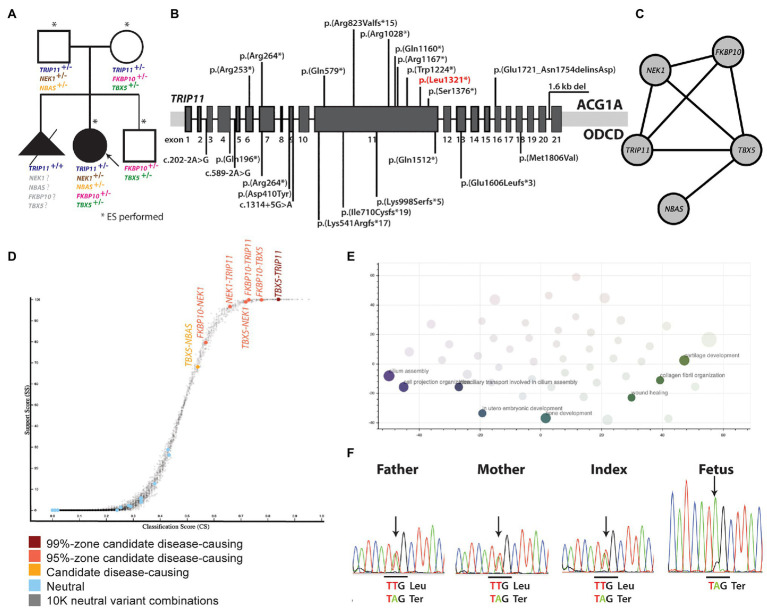
Skeletal dysplasias are often well characterized, and only a minority of the cases remain unsolved after a thorough analysis of pathogenic variants in over 400 genes that are presently known to cause monogenic skeletal diseases. Here, we describe an 11-year-old Finnish girl, born to unrelated healthy parents, who had severe short stature and a phenotype similar to odontochondrodysplasia (ODCD), a monogenic skeletal dysplasia caused by biallelic TRIP11 variants. The family had previously lost a fetus due to severe skeletal dysplasia. Exome sequencing and bioinformatic analysis revealed an oligogenic inheritance of a heterozygous nonsense mutation in TRIP11 and four likely pathogenic missense variants in FKBP10, TBX5, NEK1, and NBAS in the index patient. Interestingly, all these genes except TBX5 are known to cause skeletal dysplasia in an autosomal recessive manner. In contrast, the fetus was found homozygous for the TRIP11 mutation, and achondrogenesis type IA diagnosis was, thus, molecularly confirmed, indicating two different skeletal dysplasia forms in the family. To the best of our knowledge, this is the first report of an oligogenic inheritance model of a skeletal dysplasia in a Finnish family. Our findings may have implications for genetic counseling and for understanding the yet unsolved cases of rare skeletal dysplasias.
Keywords: TRIP11; achondrogenesis type IA; odontochondrodysplasia; oligogenic inheritance; short stature.
Copyright © 2021 Costantini, Valta, Suomi, Mäkitie and Taylan.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures
References
-
- Aoki T., Ichimura S., Itoh A., Kuramoto M., Shinkawa T., Isobe T., et al. . (2009). Identification of the neuroblastoma-amplified gene product as a component of the syntaxin 18 complex implicated in Golgi-to-endoplasmic reticulum retrograde transport. Mol. Biol. Cell 20, 2639–2649. 10.1091/mbc.e08-11-1104, PMID: - DOI - PMC - PubMed
-
- Barnes A. M., Cabral W. A., Weis M., Makareeva E., Mertz E. L., Leikin S., et al. . (2012). Absence of FKBP10 in recessive type XI osteogenesis imperfecta leads to diminished collagen cross-linking and reduced collagen deposition in extracellular matrix. Hum. Mutat. 33, 1589–1598. 10.1002/humu.22139, PMID: - DOI - PMC - PubMed
LinkOut - more resources
Full Text Sources