

Multiomic Analysis of Lung Tumors Defines Pathways Activated in Neuroendocrine Transformation
- PMID: 34155000
- PMCID: PMC9437746
- DOI: 10.1158/2159-8290.CD-20-1863
Multiomic Analysis of Lung Tumors Defines Pathways Activated in Neuroendocrine Transformation
Abstract
Lineage plasticity is implicated in treatment resistance in multiple cancers. In lung adenocarcinomas (LUAD) amenable to targeted therapy, transformation to small cell lung cancer (SCLC) is a recognized resistance mechanism. Defining molecular mechanisms of neuroendocrine (NE) transformation in lung cancer has been limited by a paucity of pre/posttransformation clinical samples. Detailed genomic, epigenomic, transcriptomic, and protein characterization of combined LUAD/SCLC tumors, as well as pre/posttransformation samples, supports that NE transformation is primarily driven by transcriptional reprogramming rather than mutational events. We identify genomic contexts in which NE transformation is favored, including frequent loss of the 3p chromosome arm. We observed enhanced expression of genes involved in the PRC2 complex and PI3K/AKT and NOTCH pathways. Pharmacologic inhibition of the PI3K/AKT pathway delayed tumor growth and NE transformation in an EGFR-mutant patient-derived xenograft model. Our findings define a novel landscape of potential drivers and therapeutic vulnerabilities of NE transformation in lung cancer.
Significance: The difficulty in collection of transformation samples has precluded the performance of molecular analyses, and thus little is known about the lineage plasticity mechanisms leading to LUAD-to-SCLC transformation. Here, we describe biological pathways dysregulated upon transformation and identify potential predictors and potential therapeutic vulnerabilities of NE transformation in the lung. See related commentary by Meador and Lovly, p. 2962. This article is highlighted in the In This Issue feature, p. 2945.
©2021 American Association for Cancer Research.
Conflict of interest statement
CONFLICT OF INTEREST
CAID receives research support from Bristol Myers Squibb.
CMR has consulted regarding oncology drug development with AbbVie, Amgen, Ascentage, Astra Zeneca, Bicycle, Celgene, Daiichi Sankyo, Genentech/Roche, Ipsen, Jazz, Lilly, Pfizer, PharmaMar, Syros, and Vavotek. CMR serves on the scientific advisory boards of Bridge Medicines, Earli, and Harpoon Therapeutics.
Figures
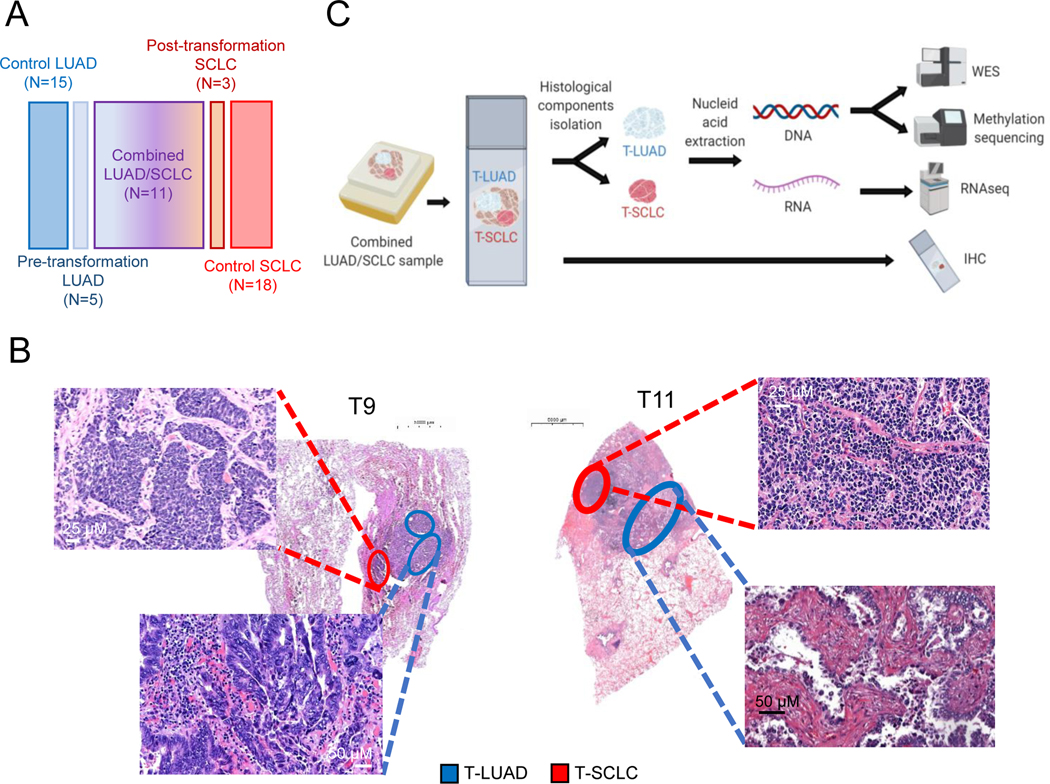
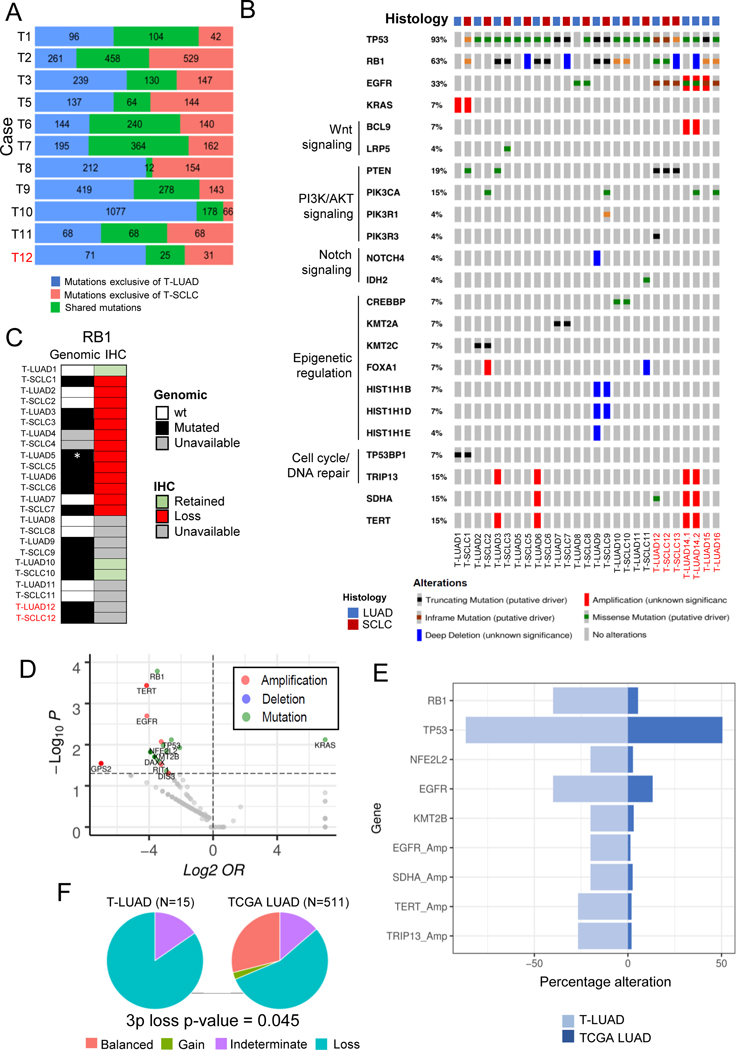
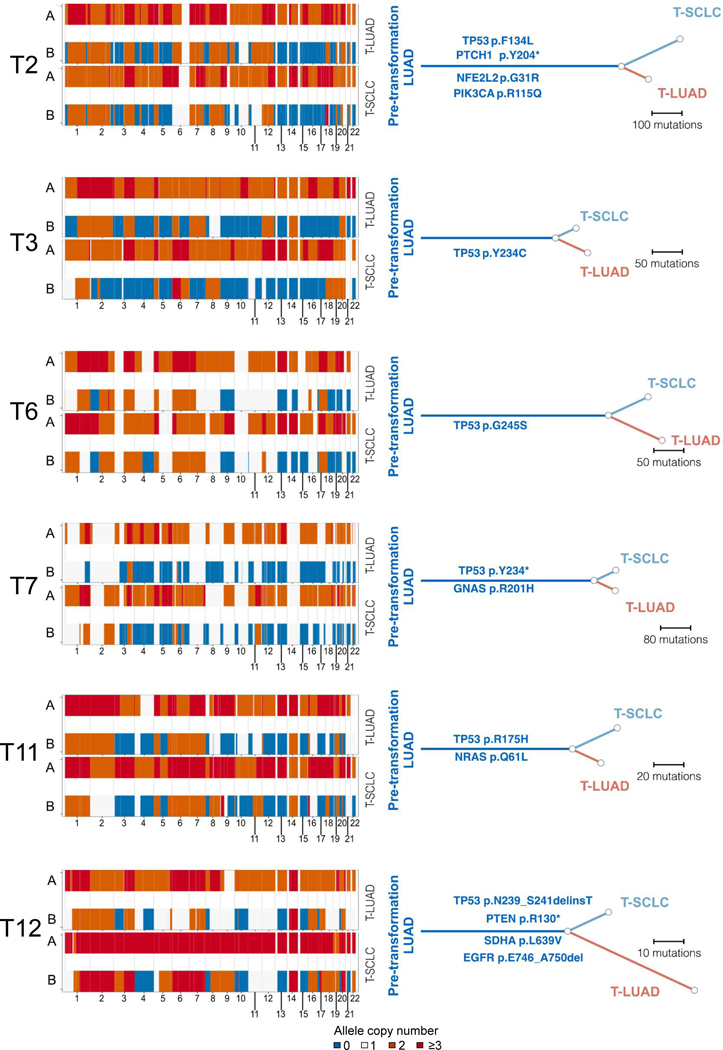
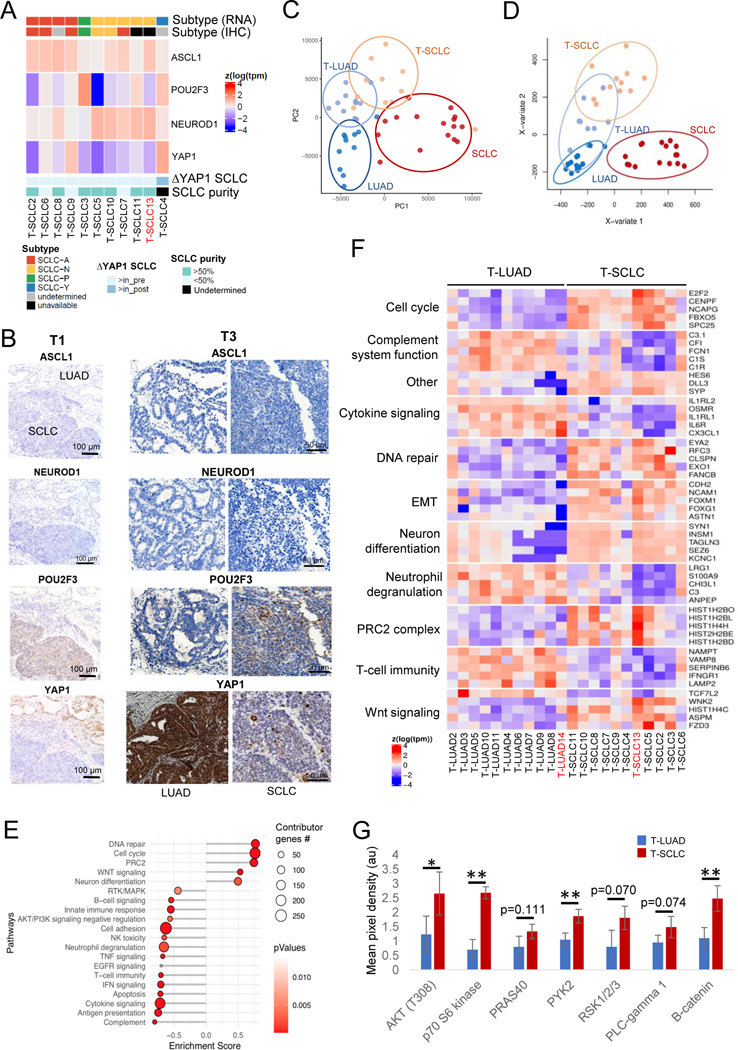
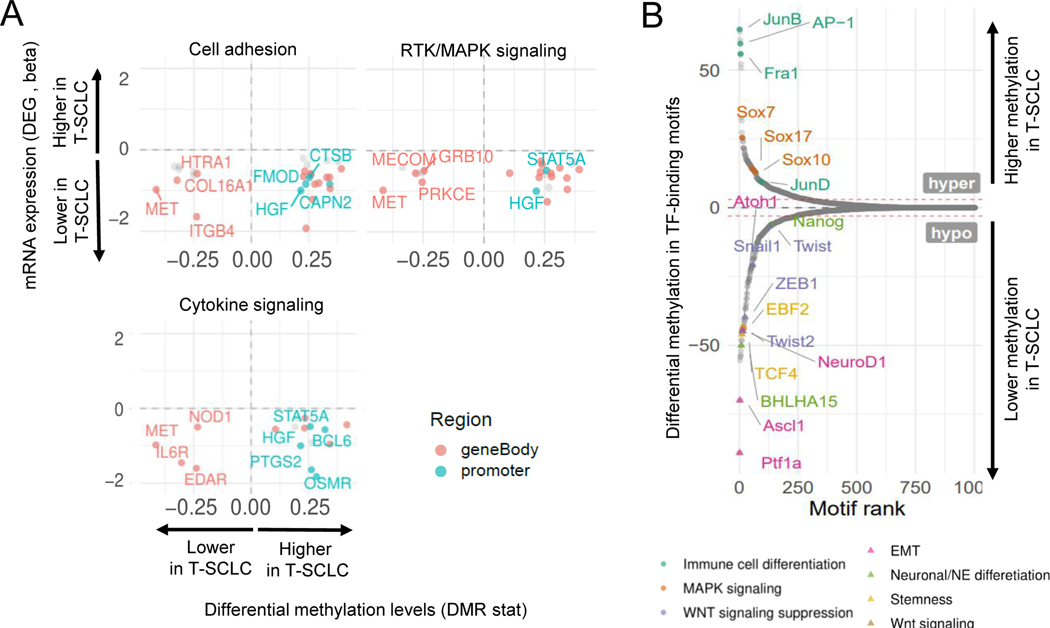
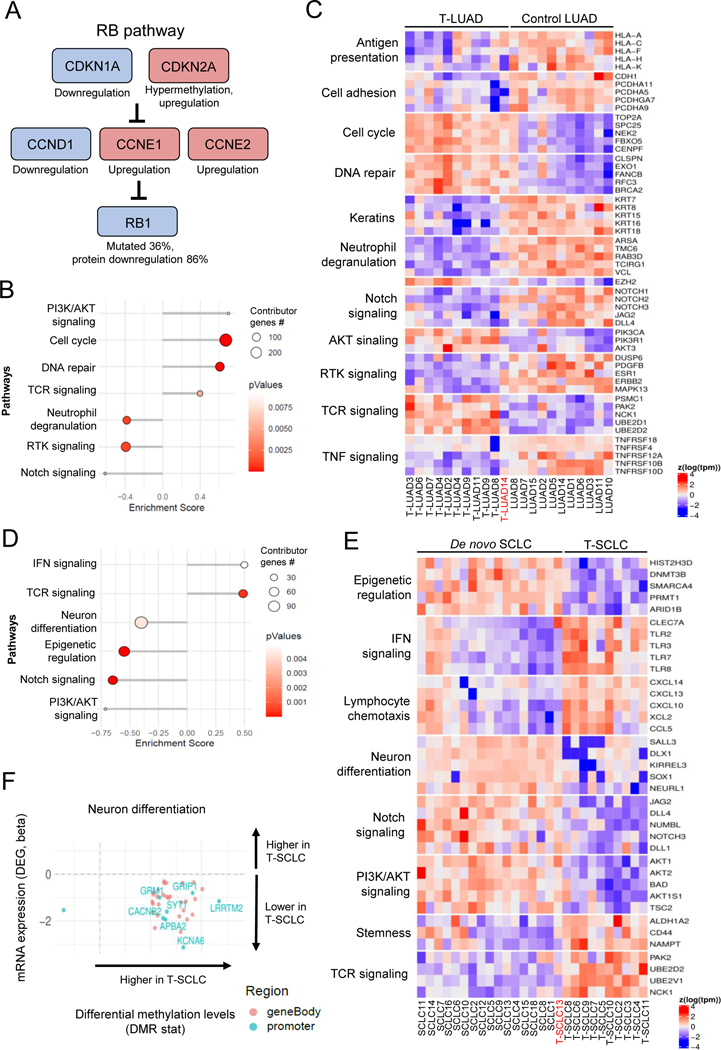
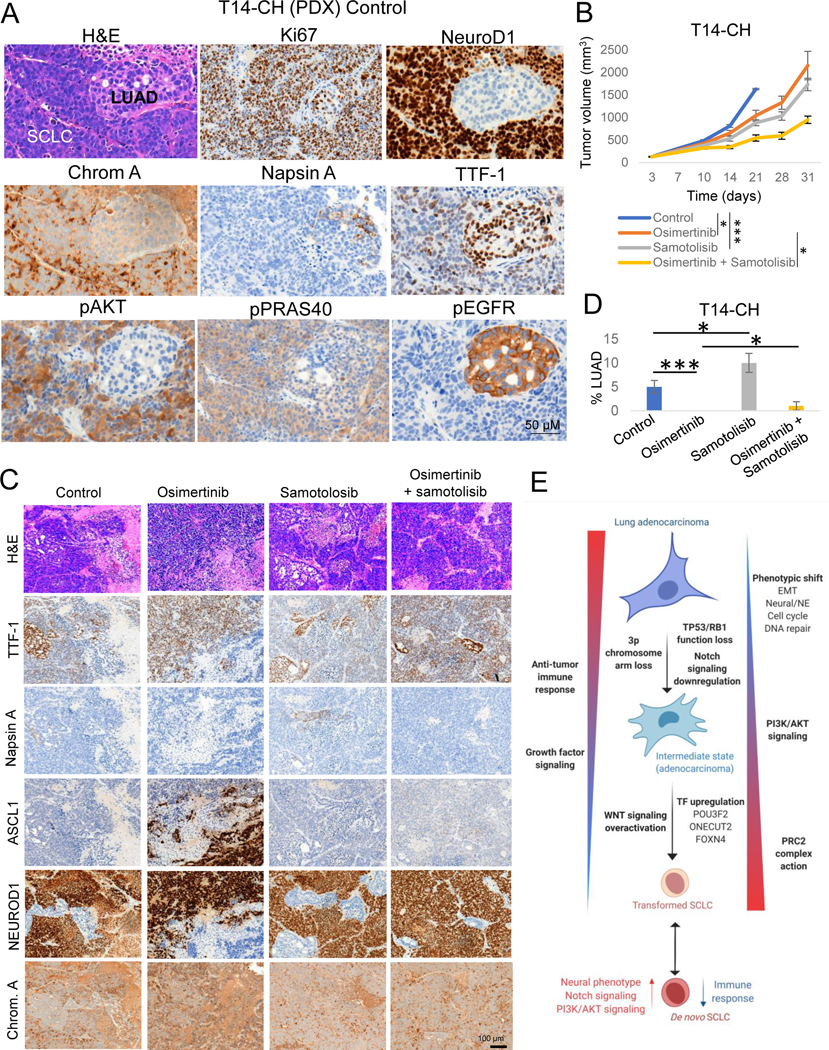
Comment in
-
A Tale of Two Histologies: Dissecting the Biology of Lineage Transformation in Lung Cancer.Cancer Discov. 2021 Dec 1;11(12):2962-2964. doi: 10.1158/2159-8290.CD-21-1222. Cancer Discov. 2021. PMID: 35363868 Free PMC article.
References
-
- Miyoshi Y, Uemura H, Kitami K, Satomi Y, Kubota Y, Hosaka M. Neuroendocrine differentiated small cell carcinoma presenting as recurrent prostate cancer after androgen deprivation therapy. BJU Int. 2001;88:982–3. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
