

Mitochondrial Disorders
- PMID: 34158150
- PMCID: PMC8830351
- DOI: 10.3238/arztebl.m2021.0251
Mitochondrial Disorders
Abstract
Background: Mitochondrial disorders are among the most common heritable diseases, with an overall lifetime risk of approximately one in 1500. Nonetheless, their diagnosis is often missed because of their extreme phenotypic and genotypic heterogeneity.
Methods: This review is based on publications retrieved by a selective literature search on the clinical features, genetics, pathogenesis, diagnosis, and treatment of mitochondrial diseases.
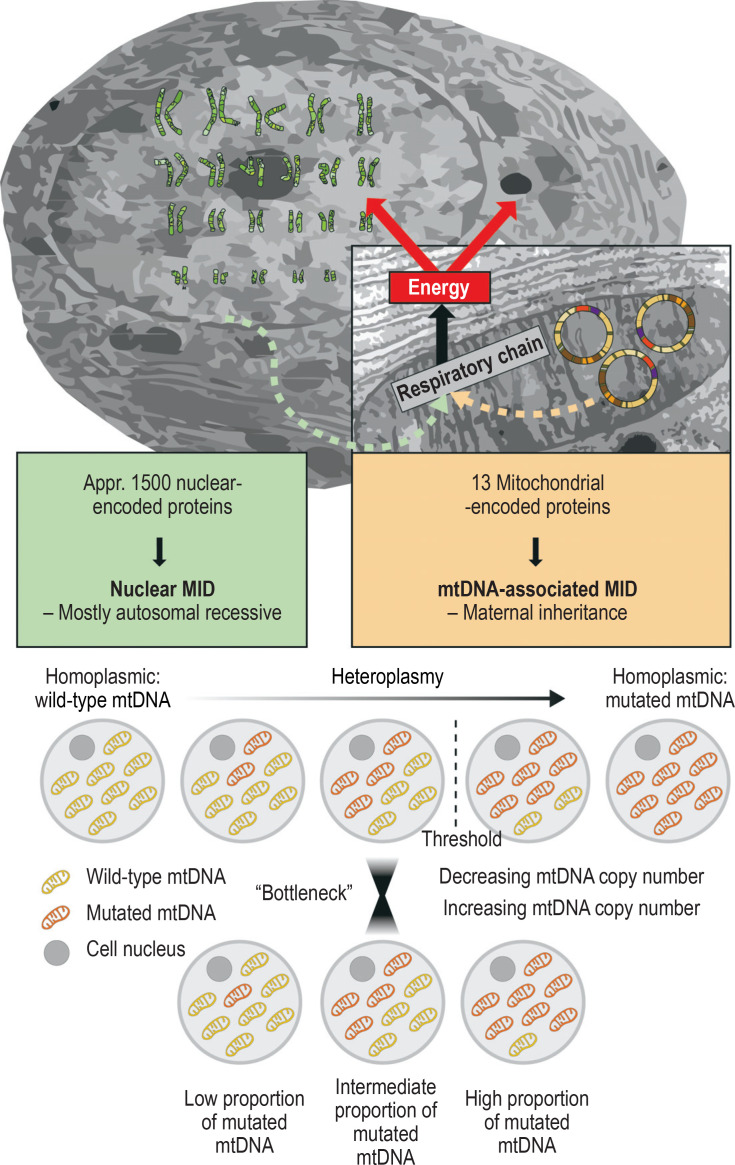
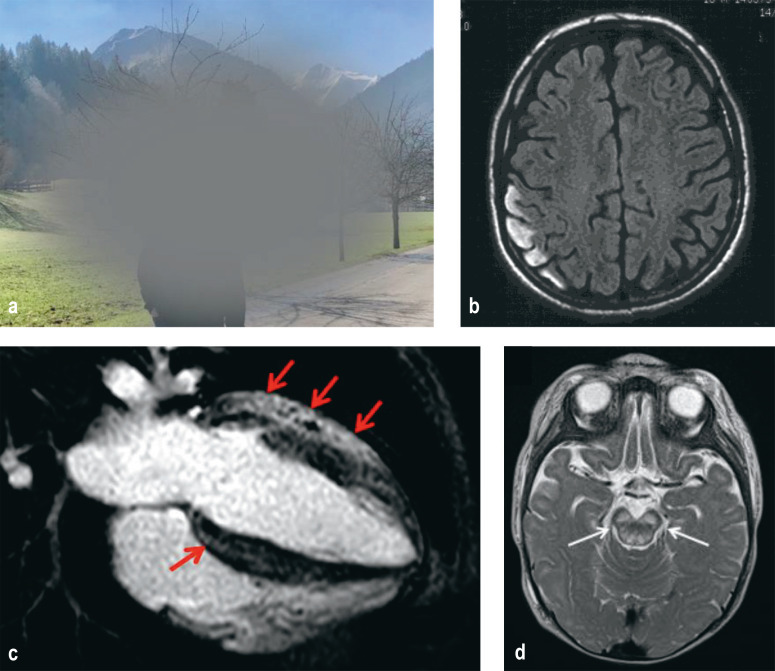
Results: Pathogenic defects of energy metabolism have been described to date in over 400 genes. Only a small number of these genes lie in the mitochondrial DNA; the corresponding diseases are either maternally inherited or of sporadic distribution. The remaining disease-associated genes are coded in nuclear DNA and cause diseases that are inherited according to Mendelian rules, mostly autosomal recessive. The most severely involved organs are generally those with the highest energy requirements, including the brain, the sensory epithelia, and the extraocular, cardiac, and skeletal musculature. Typical manifestations include epileptic seizures, stroke-like episodes, hearing loss, retinopathy, external ophthalmoparesis, exercise intolerance, and diabetes mellitus. More than two manifestations of these types should arouse suspicion of a disease of energy metabolism. The severity of mitochondrial disorders ranges from very severe disease, already evident in childhood, to relatively mild disease arising in late adulthood. The diagnosis is usually confirmed with molecular-genetic methods. Symptomatic treatment can improve patients' quality of life. The only disease-modifying treatment that has been approved to date is idebenone for the treatment of Leber hereditary optic neuropathy. Intravitreal gene therapy has also been developed for the treatment of this disease; its approval by the European Medicines Agency is pending.
Conclusion: Patients with mitochondrial diseases have highly varied manifestations and can thus present to physicians in practically any branch of medicine. A correct diagnosis is the prerequisite for genetic counseling and for the initiation of personalized treatment.
Figures

Comment in
-
Mitochondrial Disorders: Endocrine Aspects.Dtsch Arztebl Int. 2022 Apr 22;119(16):296. doi: 10.3238/arztebl.m2022.0102. Dtsch Arztebl Int. 2022. PMID: 35836344 Free PMC article. No abstract available.
-
In Reply.Dtsch Arztebl Int. 2022 Apr 22;119(16):296. doi: 10.3238/arztebl.m2022.0103. Dtsch Arztebl Int. 2022. PMID: 35836345 Free PMC article. No abstract available.
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical