

Endogenous reverse transcriptase and RNase H-mediated antiviral mechanism in embryonic stem cells
- PMID: 34158624
- PMCID: PMC8217788
- DOI: 10.1038/s41422-021-00524-7
Endogenous reverse transcriptase and RNase H-mediated antiviral mechanism in embryonic stem cells
Abstract
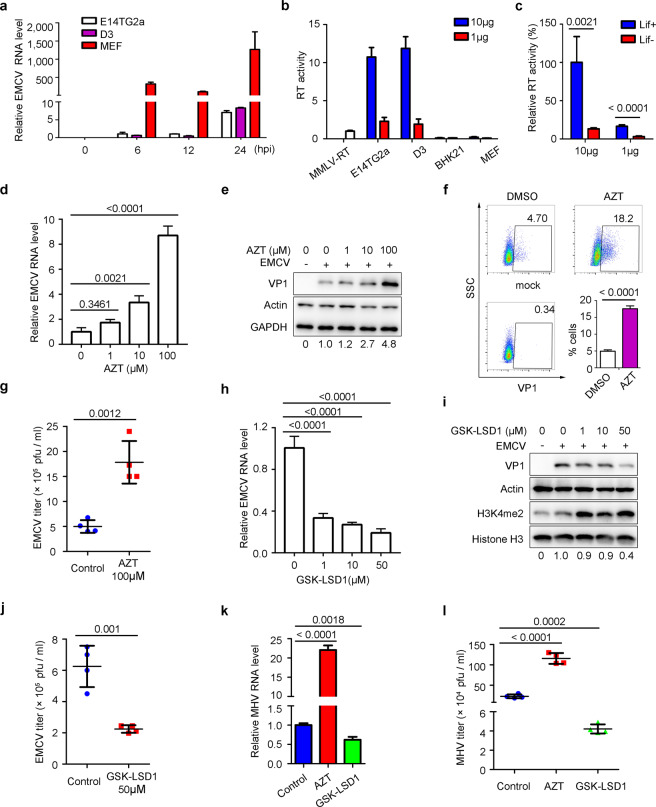
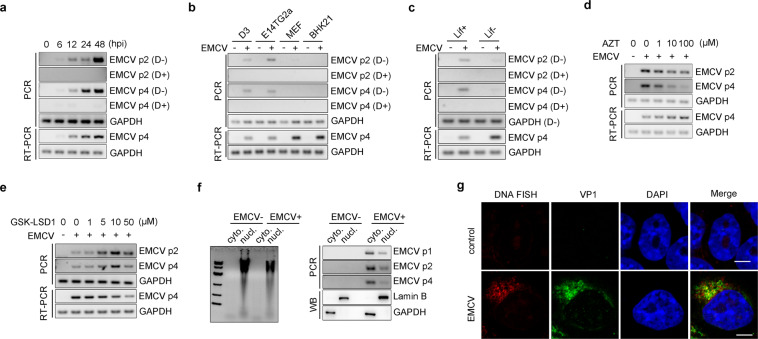
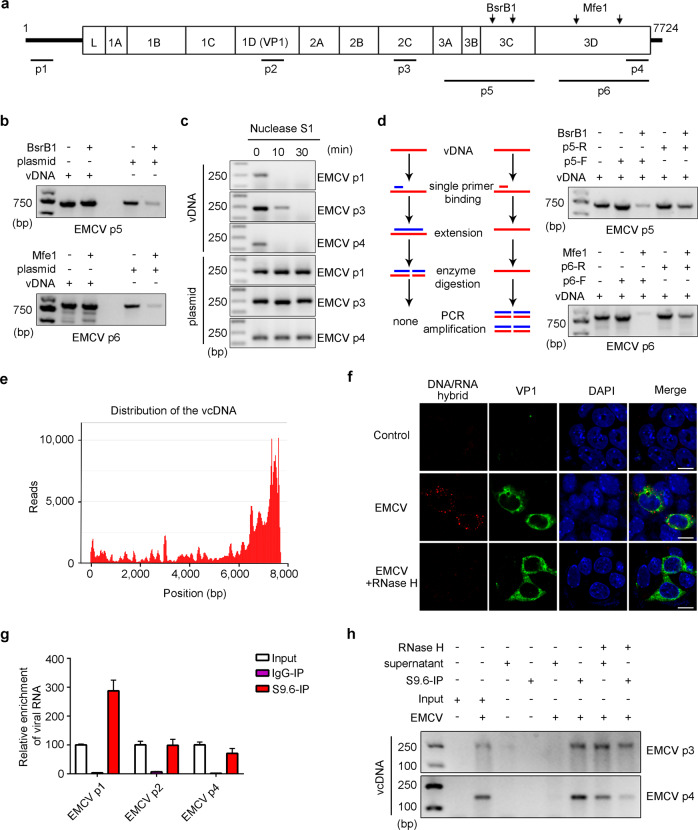
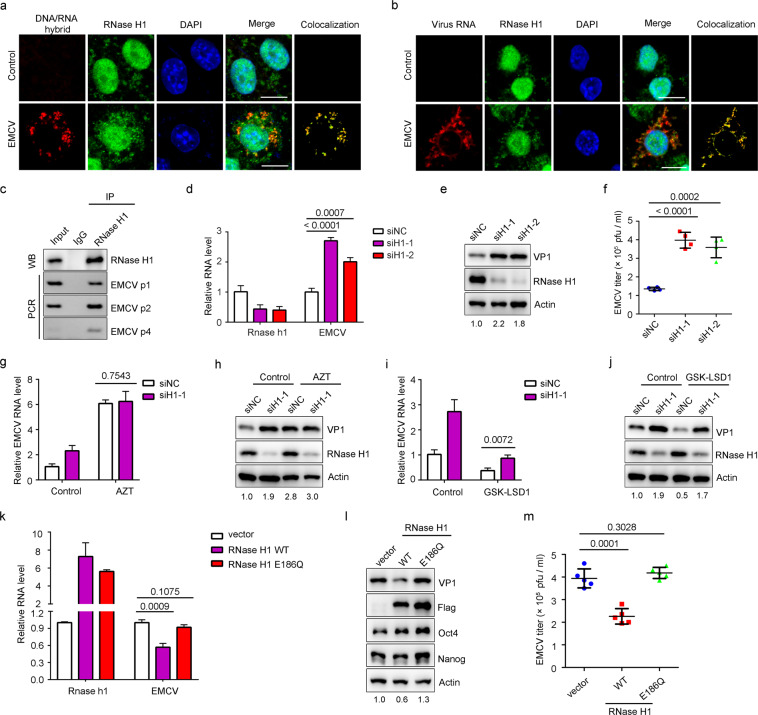
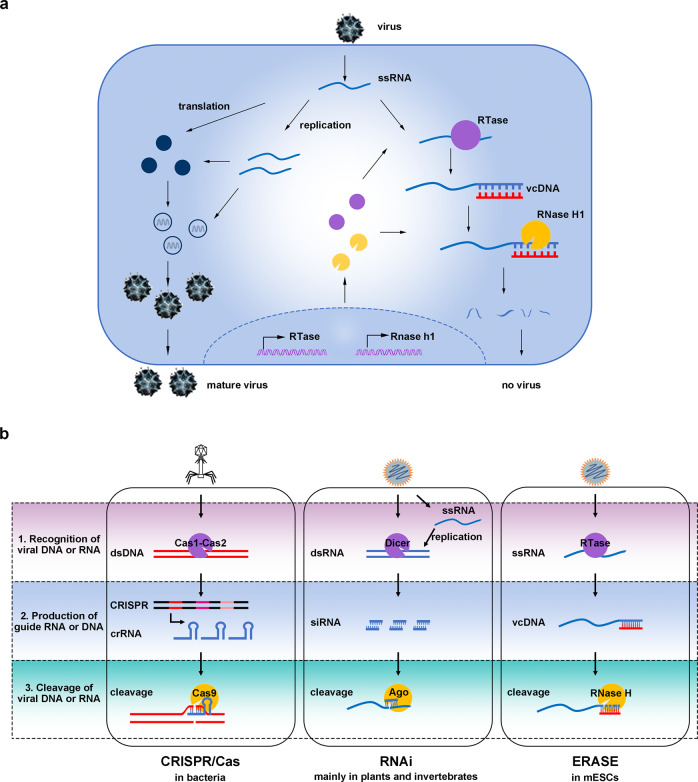
Nucleic acid-based systems play important roles in antiviral defense, including CRISPR/Cas that adopts RNA-guided DNA cleavage to prevent DNA phage infection and RNA interference (RNAi) that employs RNA-guided RNA cleavage to defend against RNA virus infection. Here, we report a novel type of nucleic acid-based antiviral system that exists in mouse embryonic stem cells (mESCs), which suppresses RNA virus infection by DNA-mediated RNA cleavage. We found that the viral RNA of encephalomyocarditis virus can be reverse transcribed into complementary DNA (vcDNA) by the reverse transcriptase (RTase) encoded by endogenous retrovirus-like elements in mESCs. The vcDNA is negative-sense single-stranded and forms DNA/RNA hybrid with viral RNA. The viral RNA in the heteroduplex is subsequently destroyed by cellular RNase H1, leading to robust suppression of viral growth. Furthermore, either inhibition of the RTase activity or depletion of endogenous RNase H1 results in the promotion of virus proliferation. Altogether, our results provide intriguing insights into the antiviral mechanism of mESCs and the antiviral function of endogenized retroviruses and cellular RNase H. Such a natural nucleic acid-based antiviral mechanism in mESCs is referred to as ERASE (endogenous RTase/RNase H-mediated antiviral system), which is an addition to the previously known nucleic acid-based antiviral mechanisms including CRISPR/Cas in bacteria and RNAi in plants and invertebrates.
© 2021. The Author(s), under exclusive licence to Center for Excellence in Molecular Cell Science, CAS.
Conflict of interest statement
The authors declare no competing interests.
Figures
Comment in
-
ERASE: a novel nucleic-acid based antiviral mechanism.Cell Res. 2021 Nov;31(11):1142-1143. doi: 10.1038/s41422-021-00568-9. Cell Res. 2021. PMID: 34526662 Free PMC article. No abstract available.
References
Publication types
MeSH terms
Substances
Grants and funding
- 32041002/National Natural Science Foundation of China (National Science Foundation of China)
- 31800151/National Natural Science Foundation of China (National Science Foundation of China)
- KQTD20180411143323605/Shenzhen Science and Technology Innovation Commission
- JSGG20200225150431472/Shenzhen Science and Technology Innovation Commission
- JCYJ20170818162249554/Shenzhen Science and Technology Innovation Commission
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
