

Loss of heterozygosity results in rapid but variable genome homogenization across yeast genetic backgrounds
- PMID: 34159898
- PMCID: PMC8245132
- DOI: 10.7554/eLife.70339
Loss of heterozygosity results in rapid but variable genome homogenization across yeast genetic backgrounds
Abstract
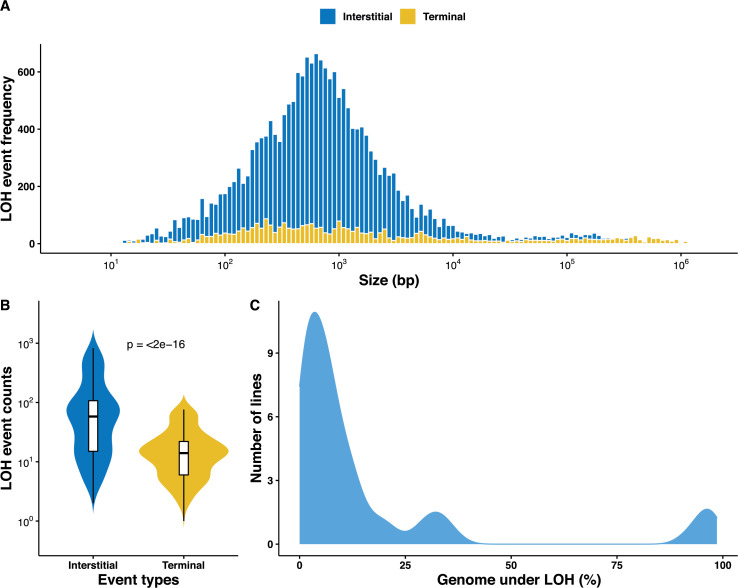
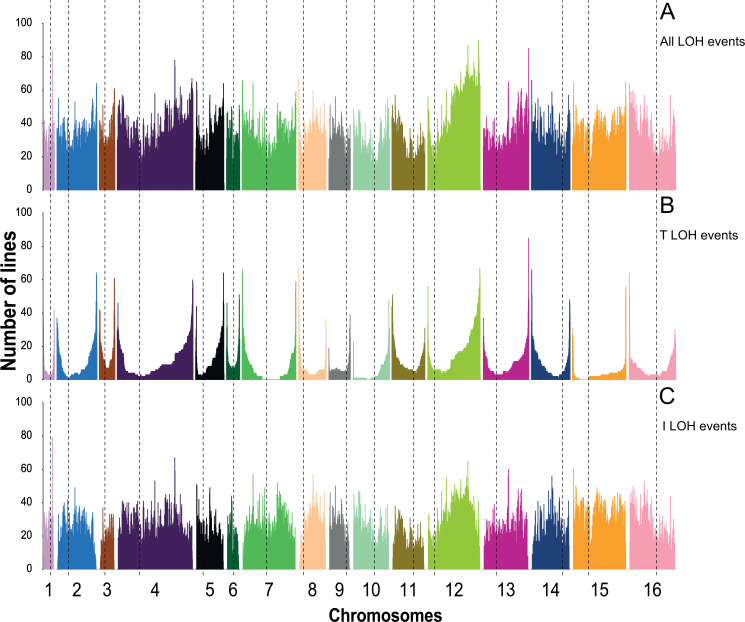
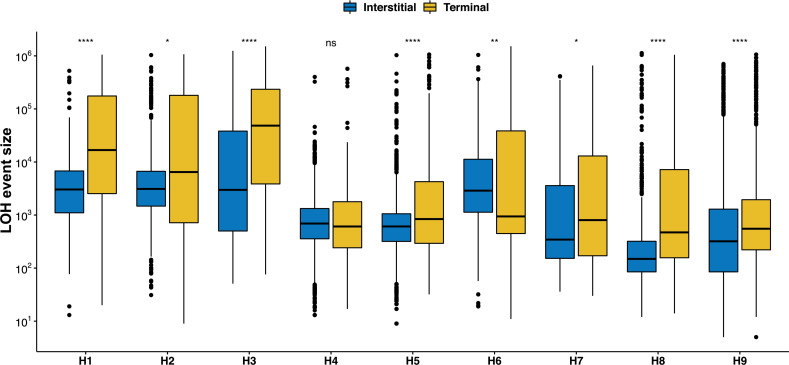
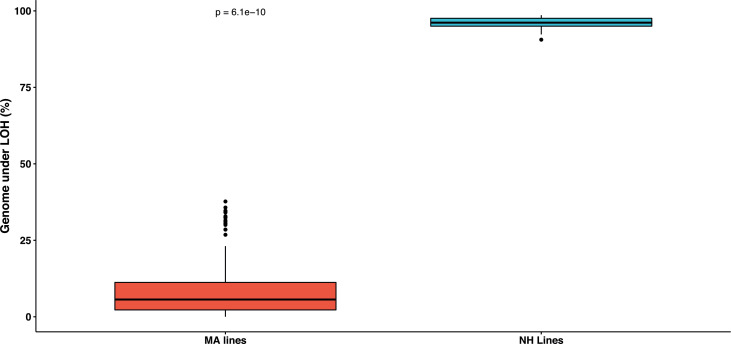
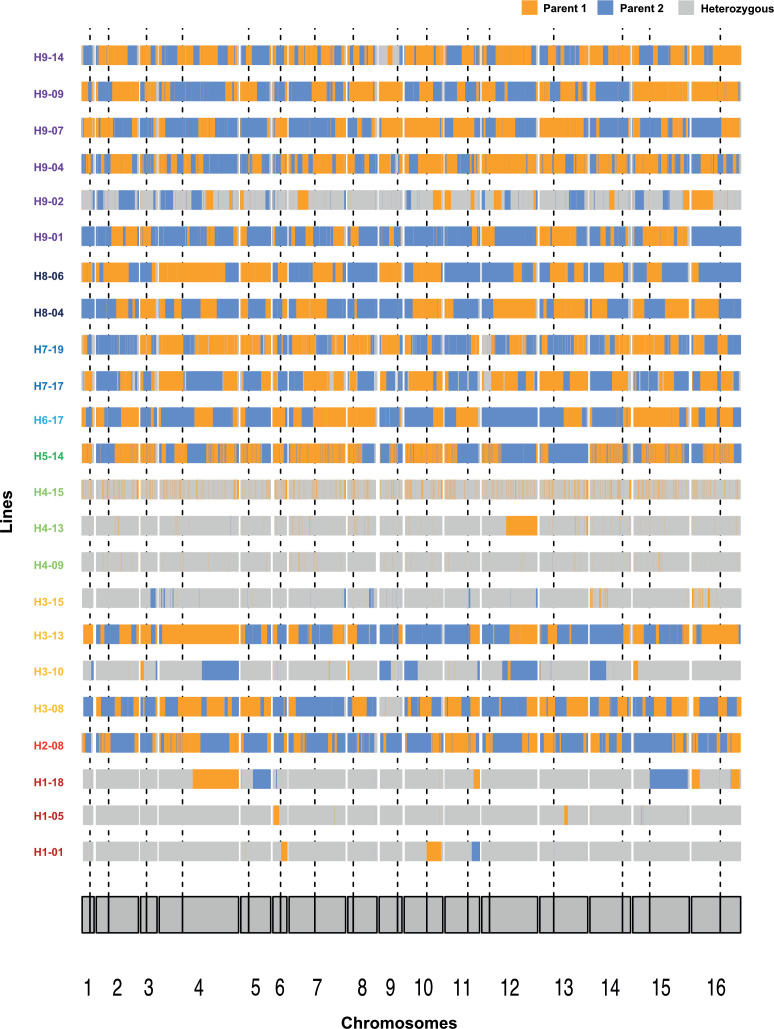
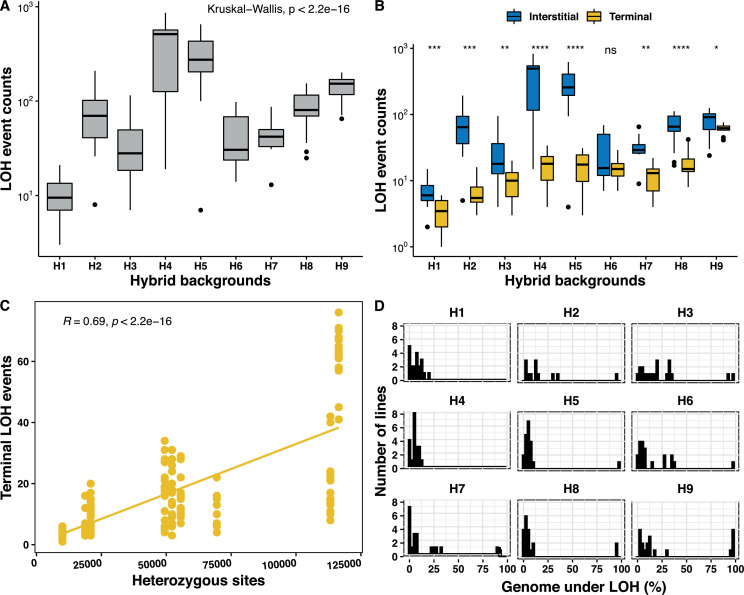
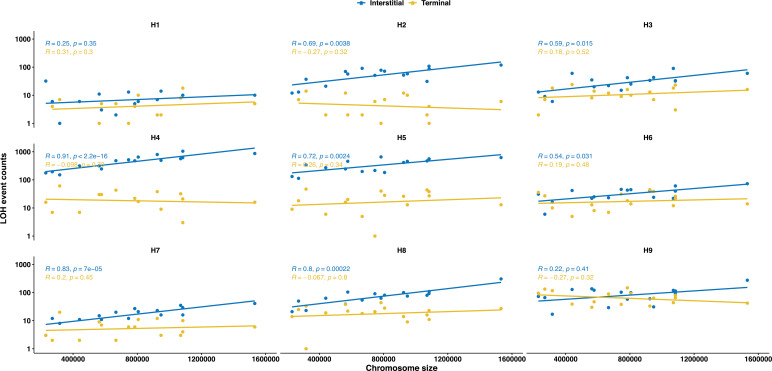
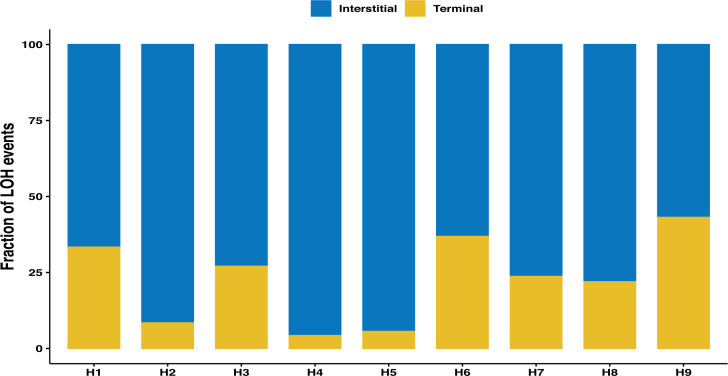
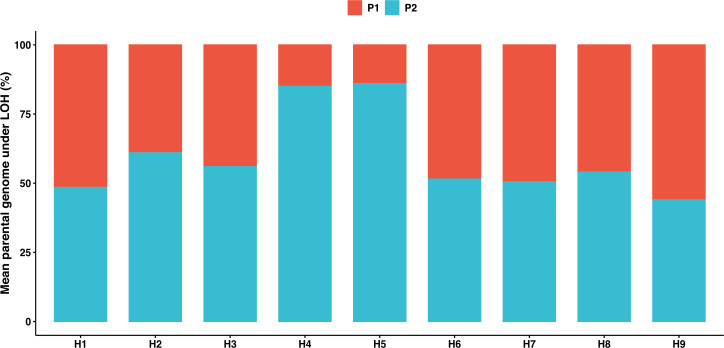
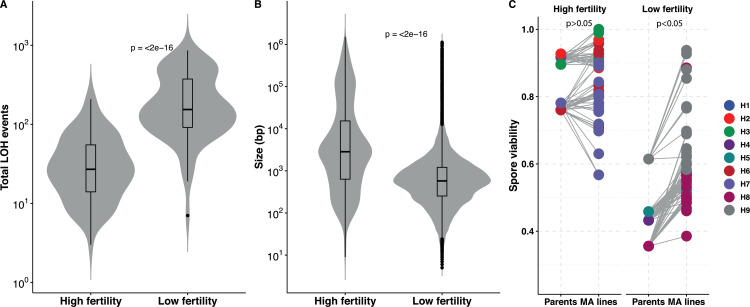
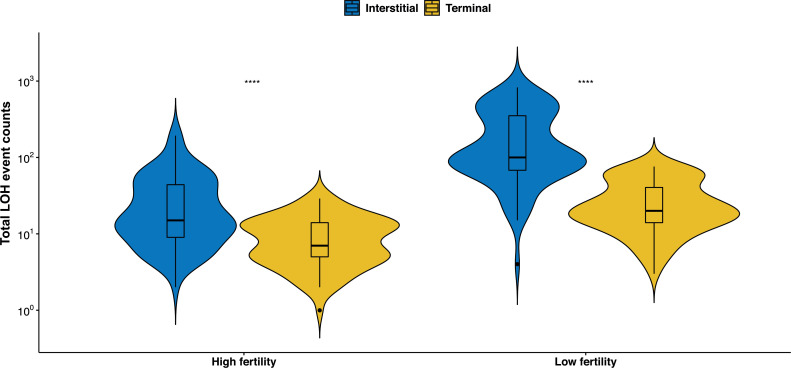
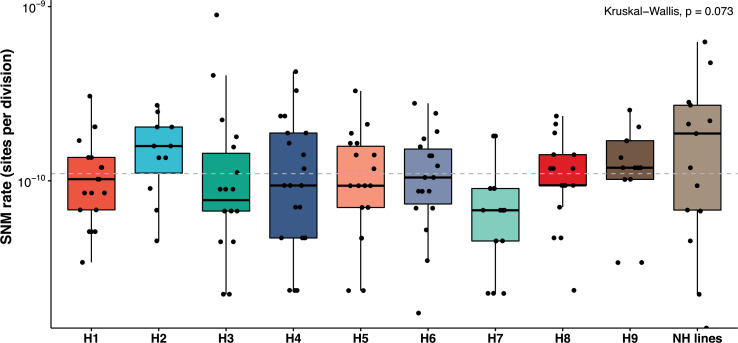
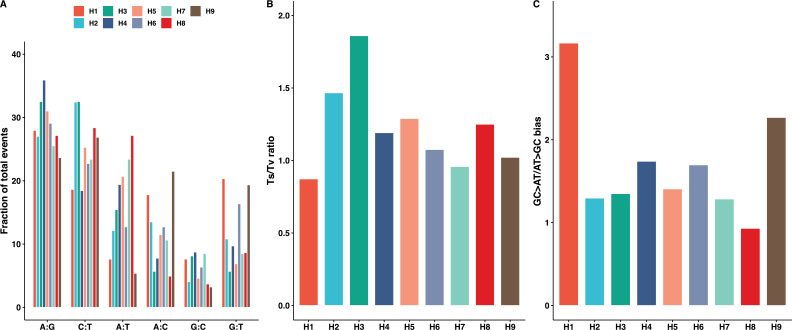
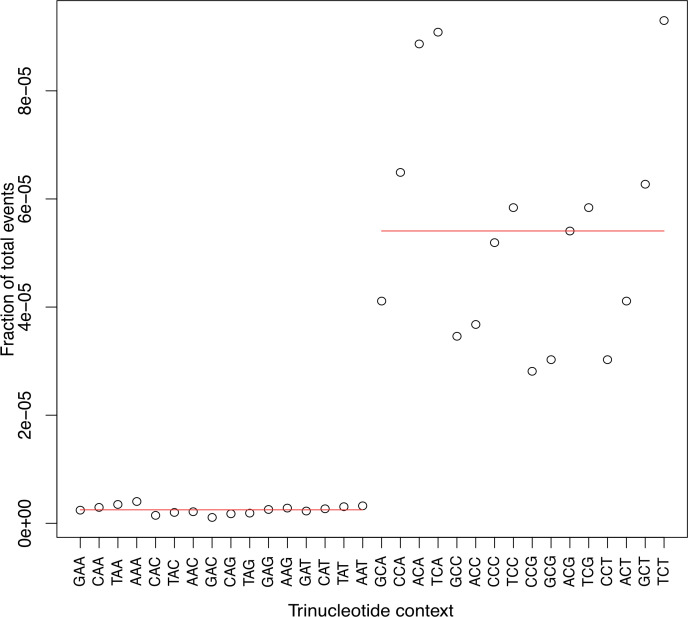
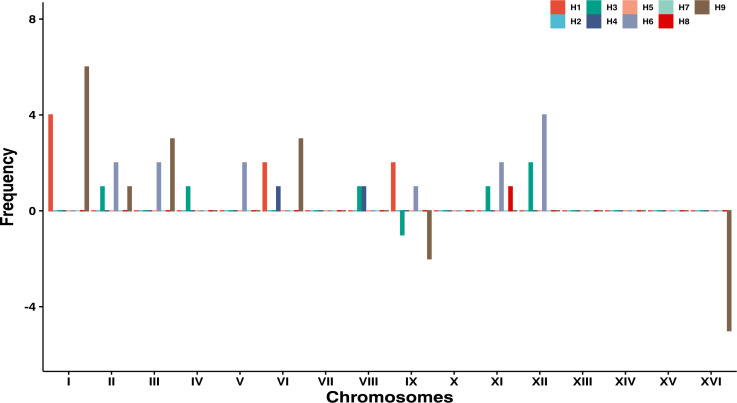
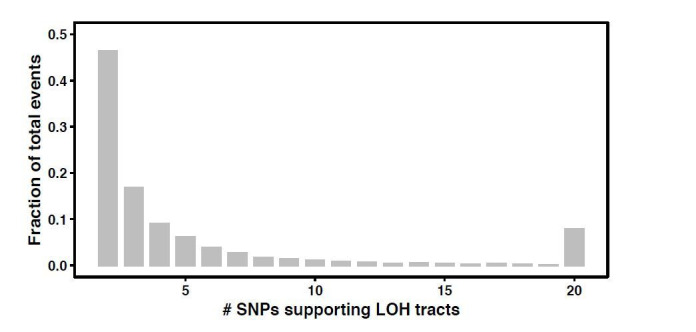
The dynamics and diversity of the appearance of genetic variants play an essential role in the evolution of the genome and the shaping of biodiversity. Recent population-wide genome sequencing surveys have highlighted the importance of loss of heterozygosity (LOH) events and have shown that they are a neglected part of the genetic diversity landscape. To assess the extent, variability, and spectrum, we explored the accumulation of LOH events in 169 heterozygous diploid Saccharomyces cerevisiae mutation accumulation lines across nine genetic backgrounds. In total, we detected a large set of 22,828 LOH events across distinct genetic backgrounds with a heterozygous level ranging from 0.1% to 1%. LOH events are very frequent with a rate consistently much higher than the mutation rate, showing their importance for genome evolution. We observed that the interstitial LOH (I-LOH) events, resulting in internal short LOH tracts, were much frequent (n = 19,660) than the terminal LOH (T-LOH) events, that is, tracts extending to the end of the chromosome (n = 3168). However, the spectrum, the rate, and the fraction of the genome under LOH vary across genetic backgrounds. Interestingly, we observed that the more the ancestors were heterozygous, the more they accumulated T-LOH events. In addition, frequent short I-LOH tracts are a signature of the lines derived from hybrids with low spore fertility. Finally, we found lines showing almost complete homozygotization during vegetative progression. Overall, our results highlight that the variable dynamics of the LOH accumulation across distinct genetic backgrounds might lead to rapid differential genome evolution during vegetative growth.
Keywords: S. cerevisiae; genetic diverstity; genetics; genomics; loss of heterozygosity; mitotic recombination; mutation accumulation; spore fertility.
© 2021, Dutta et al.
Conflict of interest statement
AD, FD, JS No competing interests declared
Figures

Similar articles
-
Genome Dynamics of Hybrid Saccharomyces cerevisiae During Vegetative and Meiotic Divisions.G3 (Bethesda). 2017 Nov 6;7(11):3669-3679. doi: 10.1534/g3.117.1135. G3 (Bethesda). 2017. PMID: 28916648 Free PMC article.
-
Loss of Heterozygosity Spectrum Depends on Ploidy Level in Natural Yeast Populations.Mol Biol Evol. 2022 Nov 3;39(11):msac214. doi: 10.1093/molbev/msac214. Mol Biol Evol. 2022. PMID: 36205042 Free PMC article.
-
Loss of Heterozygosity and Base Mutation Rates Vary Among Saccharomyces cerevisiae Hybrid Strains.G3 (Bethesda). 2020 Sep 2;10(9):3309-3319. doi: 10.1534/g3.120.401551. G3 (Bethesda). 2020. PMID: 32727920 Free PMC article.
-
Loss of Heterozygosity and Its Importance in Evolution.J Mol Evol. 2023 Jun;91(3):369-377. doi: 10.1007/s00239-022-10088-8. Epub 2023 Feb 8. J Mol Evol. 2023. PMID: 36752826 Free PMC article. Review.
-
The dynamics of loss of heterozygosity events in genomes.EMBO Rep. 2025 Feb;26(3):602-612. doi: 10.1038/s44319-024-00353-w. Epub 2025 Jan 2. EMBO Rep. 2025. PMID: 39747660 Free PMC article. Review.
Cited by
-
Genome-scale patterns in the loss of heterozygosity incidence in Saccharomyces cerevisiae.Genetics. 2022 May 5;221(1):iyac032. doi: 10.1093/genetics/iyac032. Genetics. 2022. PMID: 35212738 Free PMC article.
-
Upper Bound on the Mutational Burden Imposed by a CRISPR-Cas9 Gene-Drive Element.bioRxiv [Preprint]. 2023 Nov 29:2023.11.28.569142. doi: 10.1101/2023.11.28.569142. bioRxiv. 2023. PMID: 38076841 Free PMC article. Preprint.
-
Identifying Targets of Selection in Laboratory Evolution Experiments.J Mol Evol. 2023 Jun;91(3):345-355. doi: 10.1007/s00239-023-10096-2. Epub 2023 Feb 21. J Mol Evol. 2023. PMID: 36810618 Free PMC article. Review.
-
Overdominant Mutations Restrict Adaptive Loss of Heterozygosity at Linked Loci.Genome Biol Evol. 2021 Aug 3;13(8):evab181. doi: 10.1093/gbe/evab181. Genome Biol Evol. 2021. PMID: 34363476 Free PMC article.
-
A modified fluctuation assay reveals a natural mutator phenotype that drives mutation spectrum variation within Saccharomyces cerevisiae.Elife. 2021 Sep 15;10:e68285. doi: 10.7554/eLife.68285. Elife. 2021. PMID: 34523420 Free PMC article.
References
Publication types
MeSH terms
Associated data
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
