

Single cell derived mRNA signals across human kidney tumors
- PMID: 34162837
- PMCID: PMC8222373
- DOI: 10.1038/s41467-021-23949-5
Single cell derived mRNA signals across human kidney tumors
Abstract
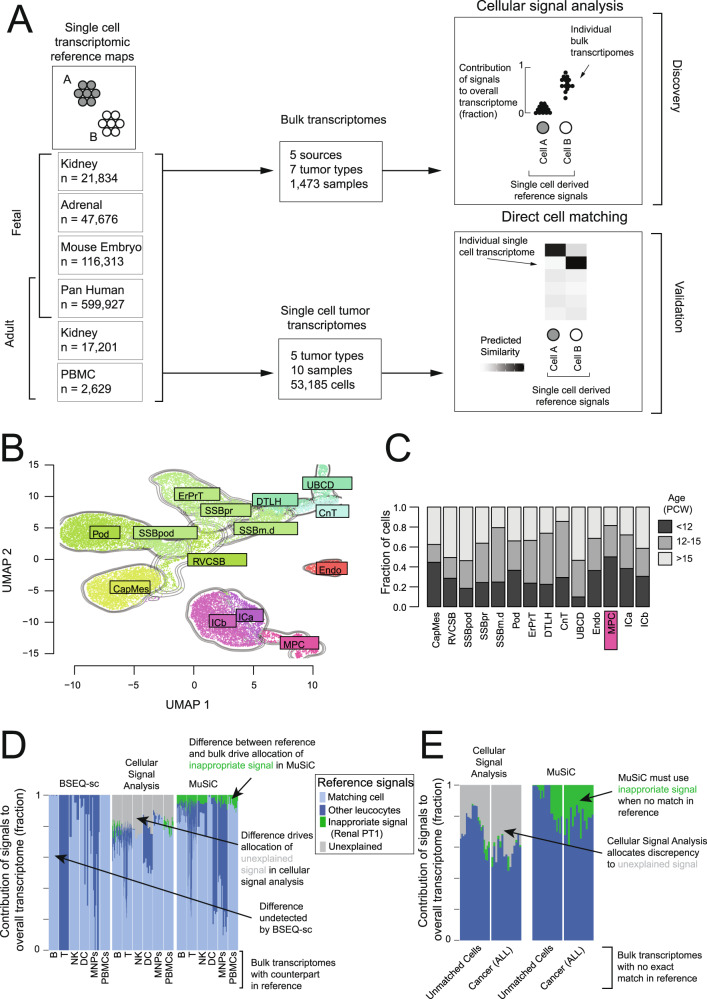
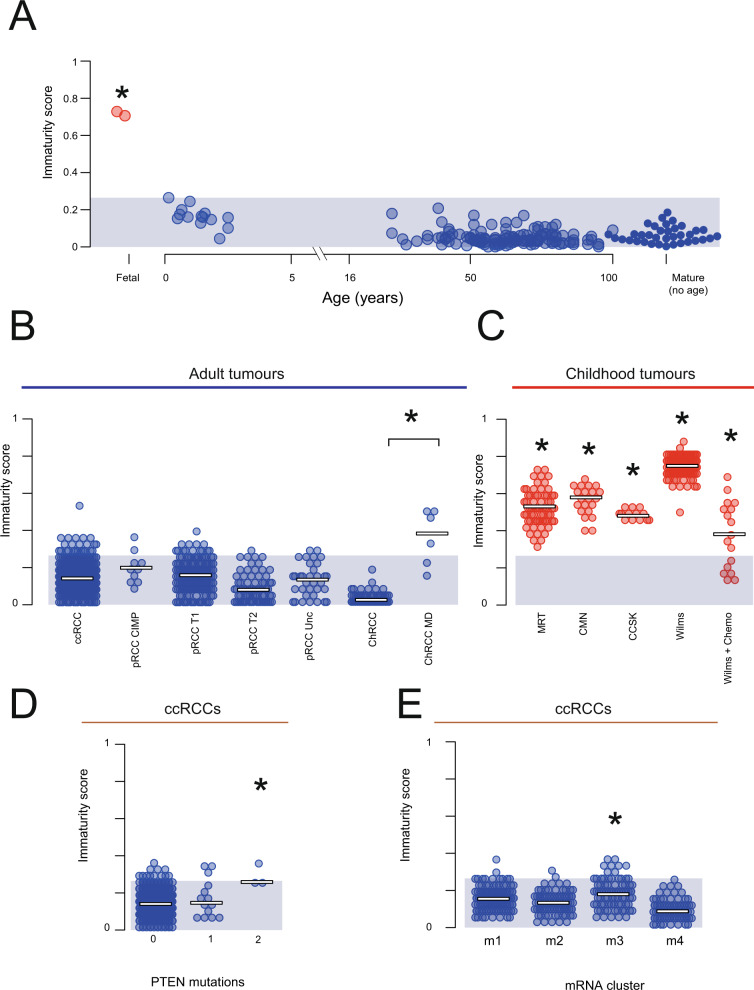
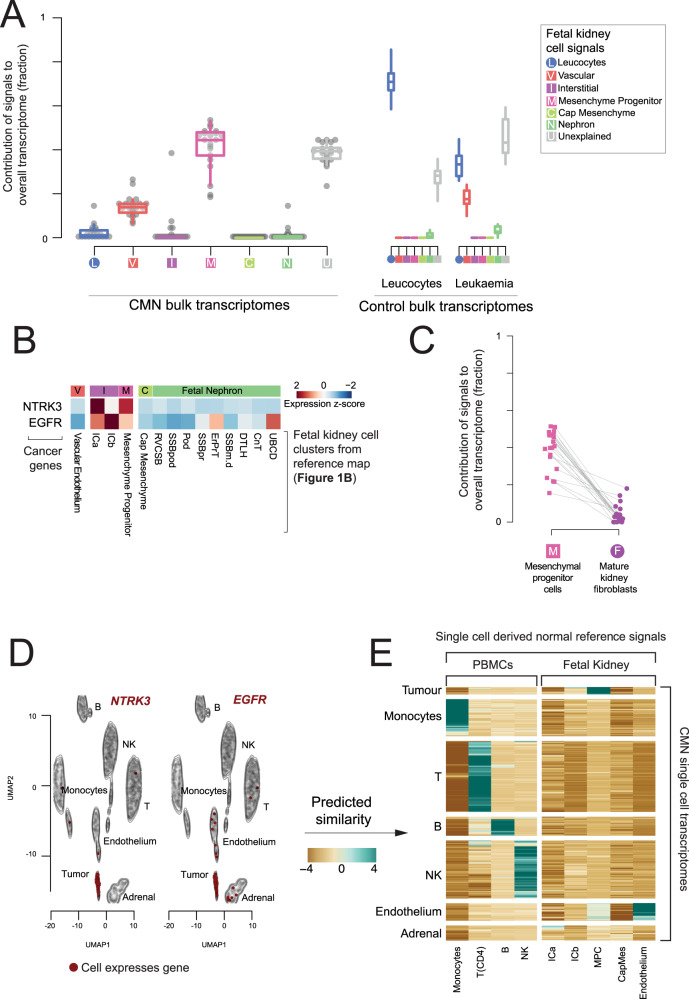
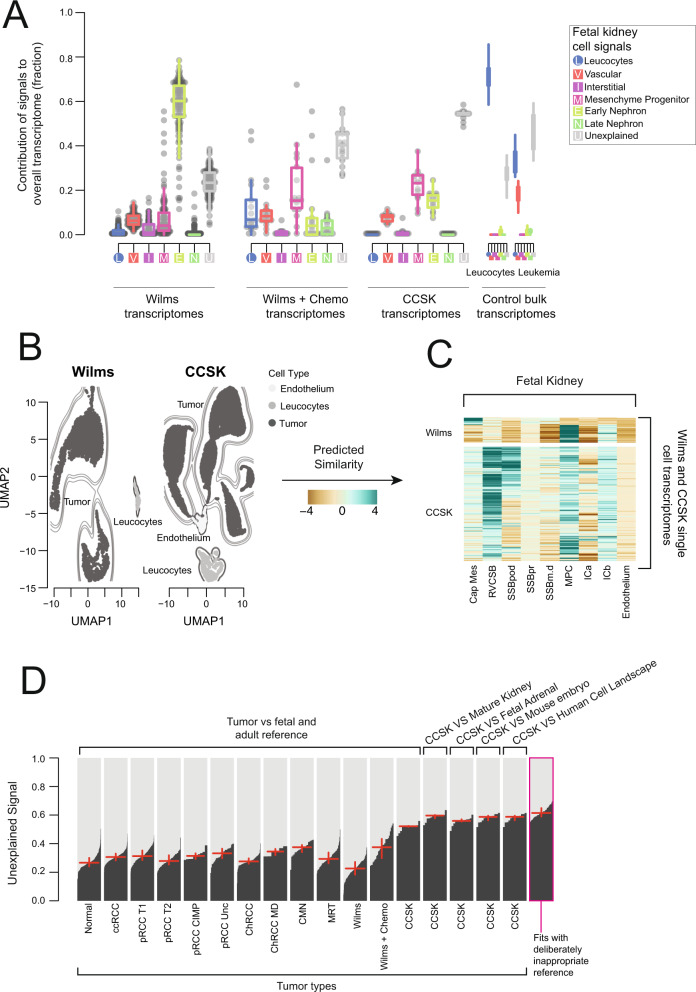
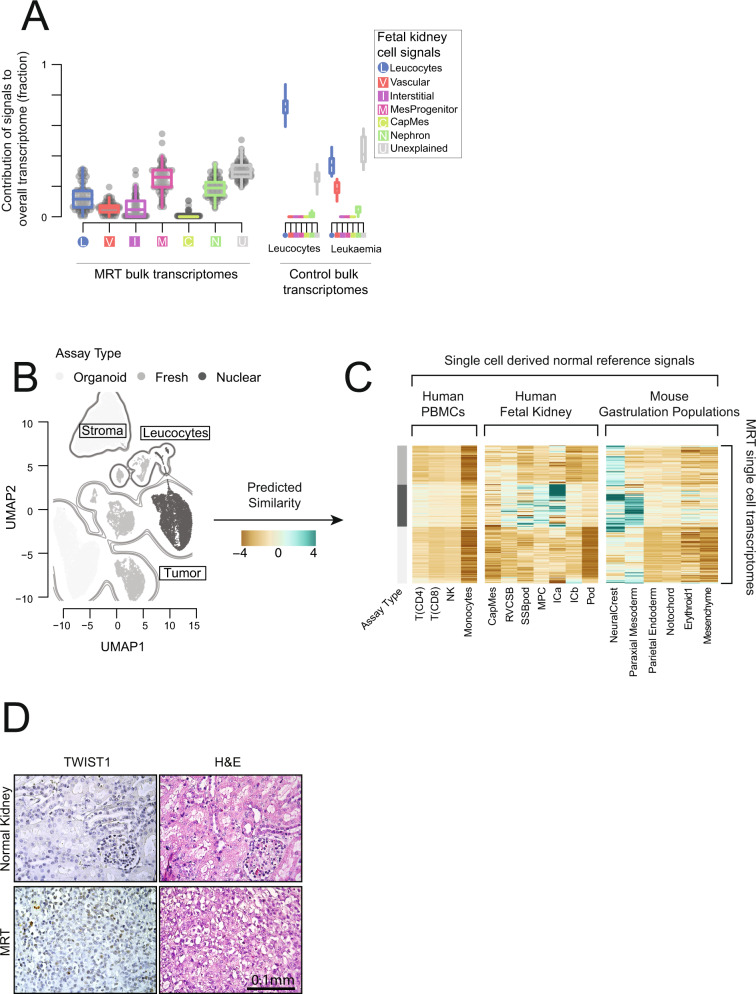
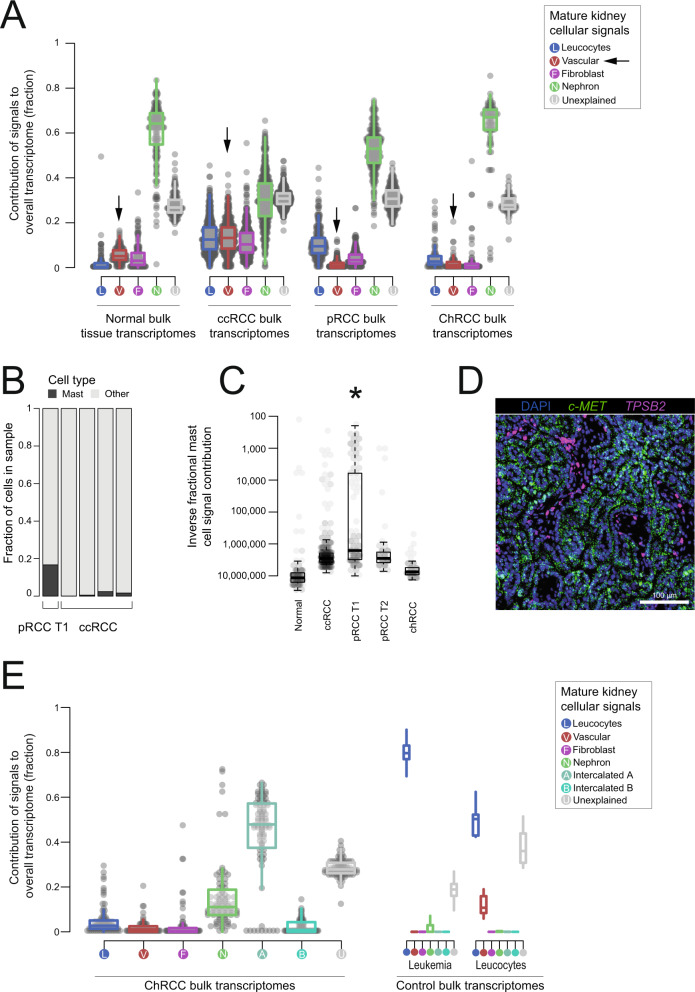
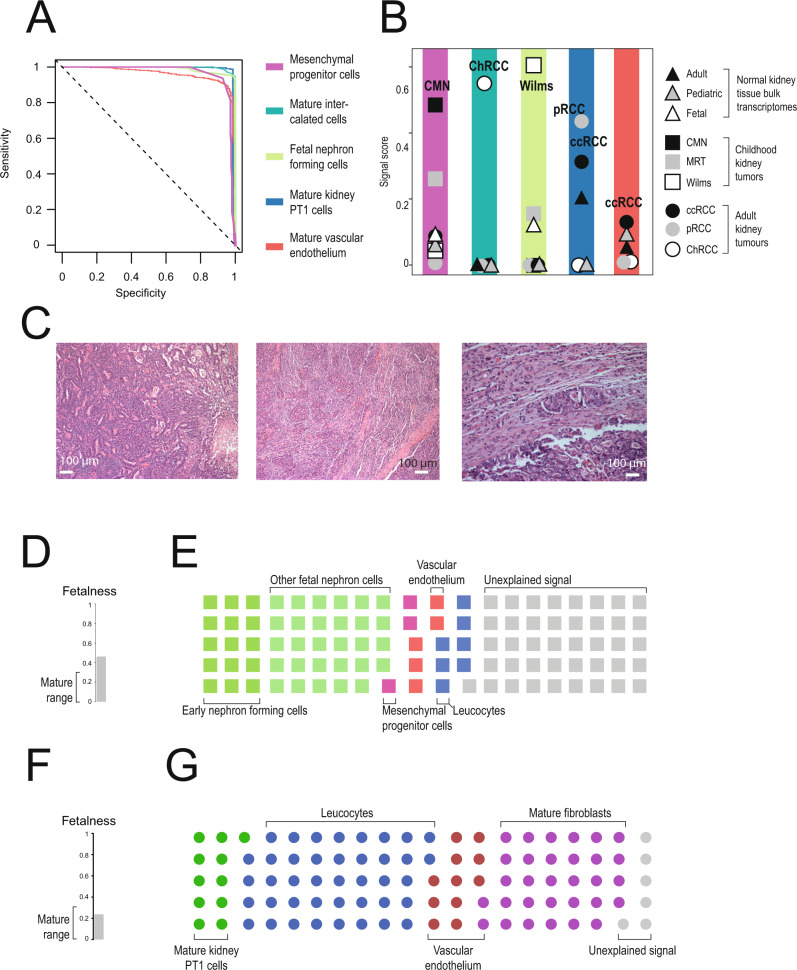
Tumor cells may share some patterns of gene expression with their cell of origin, providing clues into the differentiation state and origin of cancer. Here, we study the differentiation state and cellular origin of 1300 childhood and adult kidney tumors. Using single cell mRNA reference maps of normal tissues, we quantify reference "cellular signals" in each tumor. Quantifying global differentiation, we find that childhood tumors exhibit fetal cellular signals, replacing the presumption of "fetalness" with a quantitative measure of immaturity. By contrast, in adult cancers our assessment refutes the suggestion of dedifferentiation towards a fetal state in most cases. We find an intimate connection between developmental mesenchymal populations and childhood renal tumors. We demonstrate the diagnostic potential of our approach with a case study of a cryptic renal tumor. Our findings provide a cellular definition of human renal tumors through an approach that is broadly applicable to human cancer.
Conflict of interest statement
The authors declare no competing interests.
Figures
References
-
- A, B., P, W. & Jc, Z. SnapShot: TCGA-Analyzed Tumors. Cell173https://pubmed.ncbi.nlm.nih.gov/29625059/ (2018). - PubMed
-
- Behjati, S., Lindsay, S., Teichmann, S. A. & Haniffa, M. Mapping human development at single-cell resolution. Dev. Camb. Engl. 145, (2018). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
