

COVseq is a cost-effective workflow for mass-scale SARS-CoV-2 genomic surveillance
- PMID: 34162869
- PMCID: PMC8222401
- DOI: 10.1038/s41467-021-24078-9
COVseq is a cost-effective workflow for mass-scale SARS-CoV-2 genomic surveillance
Abstract
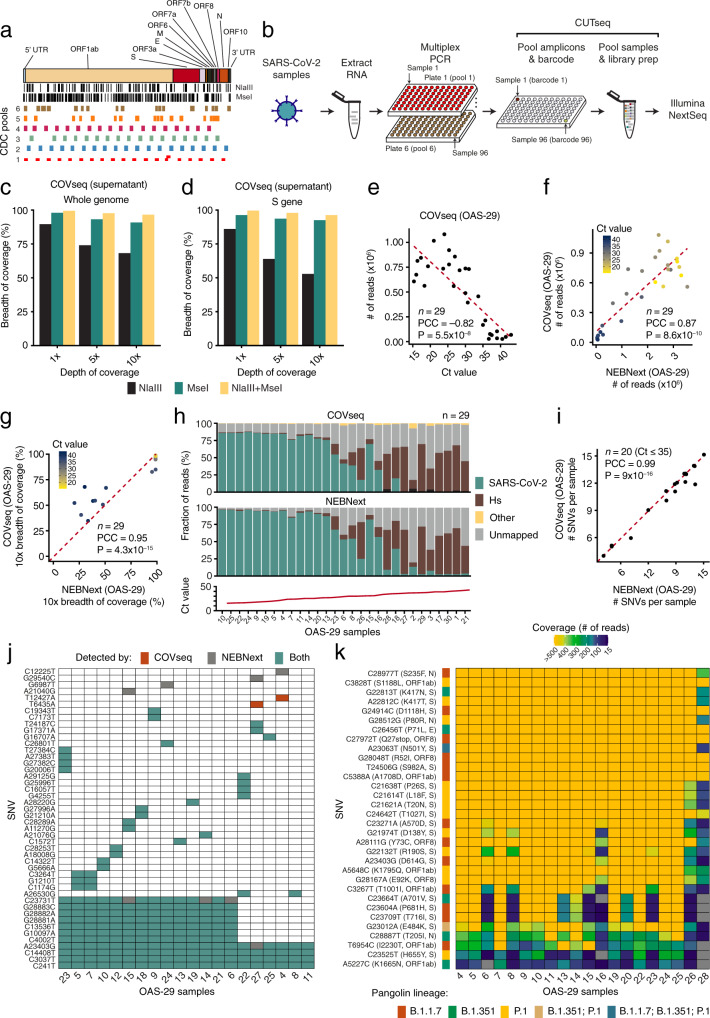
While mass-scale vaccination campaigns are ongoing worldwide, genomic surveillance of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) is critical to monitor the emergence and global spread of viral variants of concern (VOC). Here, we present a streamlined workflow-COVseq-which can be used to generate highly multiplexed sequencing libraries compatible with Illumina platforms from hundreds of SARS-CoV-2 samples in parallel, in a rapid and cost-effective manner. We benchmark COVseq against a standard library preparation method (NEBNext) on 29 SARS-CoV-2 positive samples, reaching 95.4% of concordance between single-nucleotide variants detected by both methods. Application of COVseq to 245 additional SARS-CoV-2 positive samples demonstrates the ability of the method to reliably detect emergent VOC as well as its compatibility with downstream phylogenetic analyses. A cost analysis shows that COVseq could be used to sequence thousands of samples at less than 15 USD per sample, including library preparation and sequencing costs. We conclude that COVseq is a versatile and scalable method that is immediately applicable for SARS-CoV-2 genomic surveillance and easily adaptable to other pathogens such as influenza viruses.
Conflict of interest statement
The authors declare no competing interests.
Figures



References
-
- Preliminary genomic characterisation of an emergent SARS-CoV-2 lineage in the UK defined by a novel set of spike mutations. Virologicalhttps://virological.org/t/preliminary-genomic-characterisation-of-an-eme... (2020).
-
- Tegally, H. et al. Emergence and rapid spread of a new severe acute respiratory syndrome-related coronavirus 2 (SARS-CoV-2) lineage with multiple spike mutations in South Africa. medRxiv10.1101/2020.12.21.20248640 (2020).
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
