

MULTICOM2 open-source protein structure prediction system powered by deep learning and distance prediction
- PMID: 34162922
- PMCID: PMC8222248
- DOI: 10.1038/s41598-021-92395-6
MULTICOM2 open-source protein structure prediction system powered by deep learning and distance prediction
Abstract
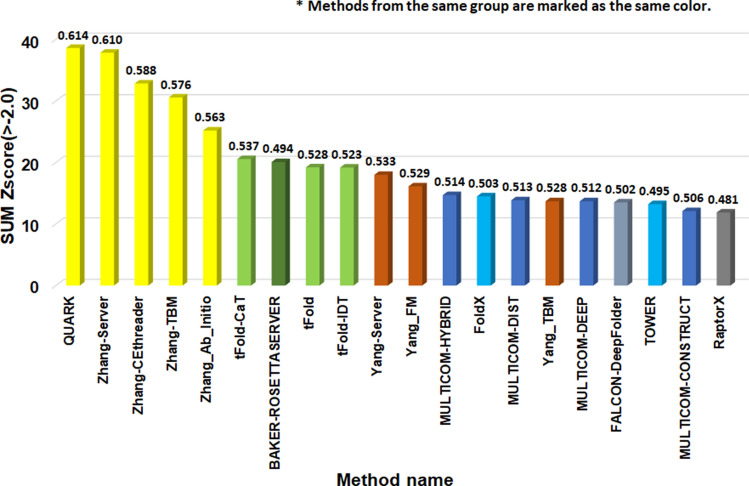
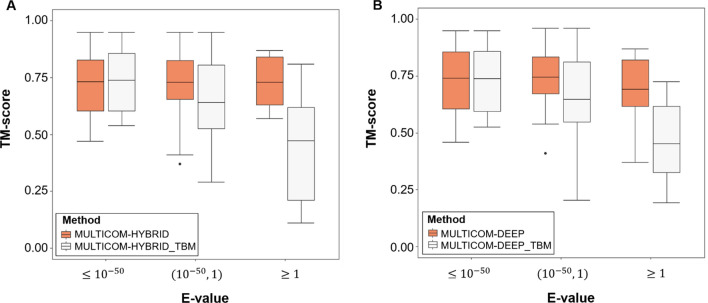
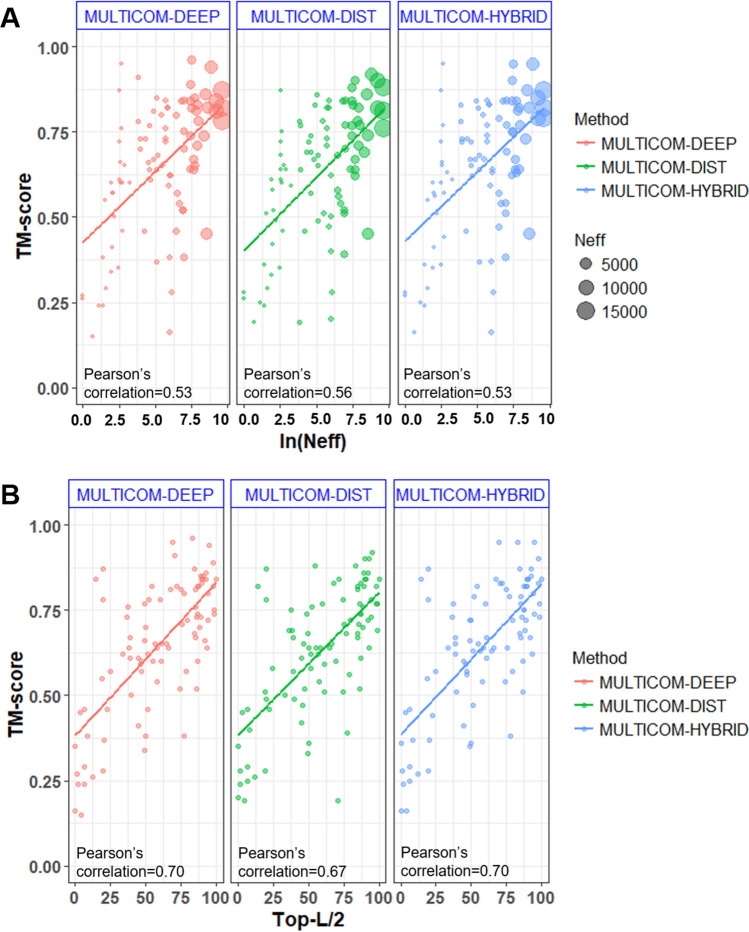
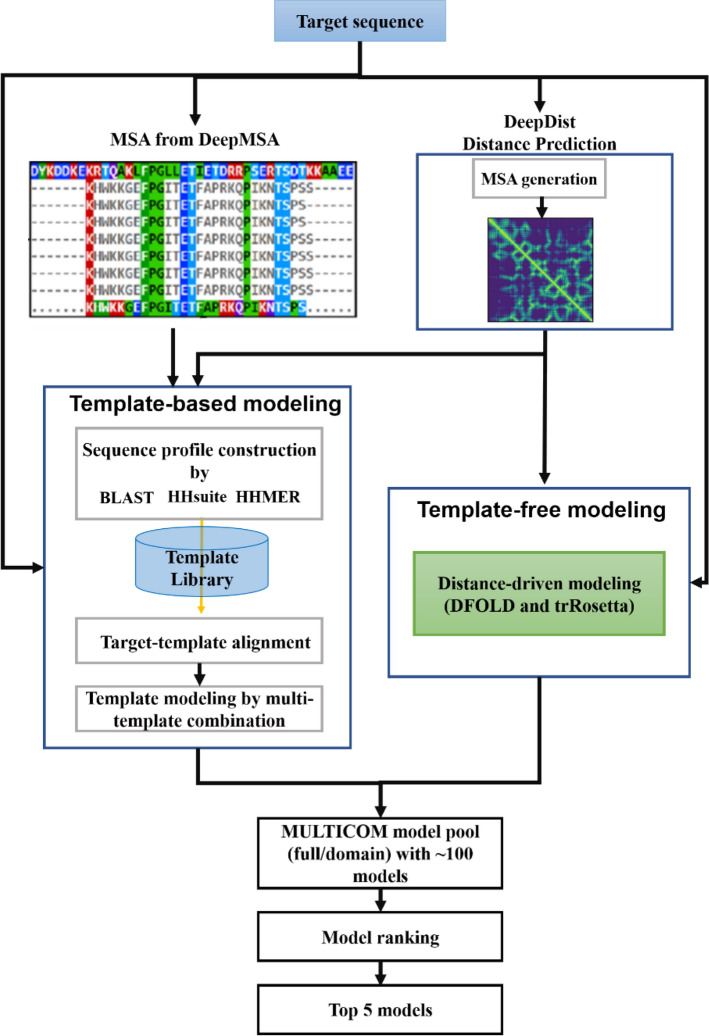
Protein structure prediction is an important problem in bioinformatics and has been studied for decades. However, there are still few open-source comprehensive protein structure prediction packages publicly available in the field. In this paper, we present our latest open-source protein tertiary structure prediction system-MULTICOM2, an integration of template-based modeling (TBM) and template-free modeling (FM) methods. The template-based modeling uses sequence alignment tools with deep multiple sequence alignments to search for structural templates, which are much faster and more accurate than MULTICOM1. The template-free (ab initio or de novo) modeling uses the inter-residue distances predicted by DeepDist to reconstruct tertiary structure models without using any known structure as template. In the blind CASP14 experiment, the average TM-score of the models predicted by our server predictor based on the MULTICOM2 system is 0.720 for 58 TBM (regular) domains and 0.514 for 38 FM and FM/TBM (hard) domains, indicating that MULTICOM2 is capable of predicting good tertiary structures across the board. It can predict the correct fold for 76 CASP14 domains (95% regular domains and 55% hard domains) if only one prediction is made for a domain. The success rate is increased to 3% for both regular and hard domains if five predictions are made per domain. Moreover, the prediction accuracy of the pure template-free structure modeling method on both TBM and FM targets is very close to the combination of template-based and template-free modeling methods. This demonstrates that the distance-based template-free modeling method powered by deep learning can largely replace the traditional template-based modeling method even on TBM targets that TBM methods used to dominate and therefore provides a uniform structure modeling approach to any protein. Finally, on the 38 CASP14 FM and FM/TBM hard domains, MULTICOM2 server predictors (MULTICOM-HYBRID, MULTICOM-DEEP, MULTICOM-DIST) were ranked among the top 20 automated server predictors in the CASP14 experiment. After combining multiple predictors from the same research group as one entry, MULTICOM-HYBRID was ranked no. 5. The source code of MULTICOM2 is freely available at https://github.com/multicom-toolbox/multicom/tree/multicom_v2.0 .
Conflict of interest statement
The authors declare no competing interests.
Figures
Similar articles
-
Improving protein tertiary structure prediction by deep learning and distance prediction in CASP14.Proteins. 2022 Jan;90(1):58-72. doi: 10.1002/prot.26186. Epub 2021 Jul 27. Proteins. 2022. PMID: 34291486 Free PMC article.
-
Improving deep learning-based protein distance prediction in CASP14.Bioinformatics. 2021 Oct 11;37(19):3190-3196. doi: 10.1093/bioinformatics/btab355. Bioinformatics. 2021. PMID: 33961009 Free PMC article.
-
The MULTICOM Protein Structure Prediction Server Empowered by Deep Learning and Contact Distance Prediction.Methods Mol Biol. 2020;2165:13-26. doi: 10.1007/978-1-0716-0708-4_2. Methods Mol Biol. 2020. PMID: 32621217
-
Toward the solution of the protein structure prediction problem.J Biol Chem. 2021 Jul;297(1):100870. doi: 10.1016/j.jbc.2021.100870. Epub 2021 Jun 11. J Biol Chem. 2021. PMID: 34119522 Free PMC article. Review.
-
Recent Progress of Protein Tertiary Structure Prediction.Molecules. 2024 Feb 13;29(4):832. doi: 10.3390/molecules29040832. Molecules. 2024. PMID: 38398585 Free PMC article. Review.
Cited by
-
Refinement of AlphaFold2 models against experimental and hybrid cryo-EM density maps.QRB Discov. 2022;3:e16. doi: 10.1017/qrd.2022.13. Epub 2022 Sep 20. QRB Discov. 2022. PMID: 37485023 Free PMC article.
-
Computational epitope-based vaccine design with bioinformatics approach; a review.Heliyon. 2025 Jan 4;11(1):e41714. doi: 10.1016/j.heliyon.2025.e41714. eCollection 2025 Jan 15. Heliyon. 2025. PMID: 39866399 Free PMC article. Review.
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
