

Molecular logic of cellular diversification in the mouse cerebral cortex
- PMID: 34163074
- PMCID: PMC9006333
- DOI: 10.1038/s41586-021-03670-5
Molecular logic of cellular diversification in the mouse cerebral cortex
Erratum in
-
Author Correction: Molecular logic of cellular diversification in the mouse cerebral cortex.Nature. 2021 Aug;596(7873):E11. doi: 10.1038/s41586-021-03797-5. Nature. 2021. PMID: 34341543 No abstract available.
Abstract
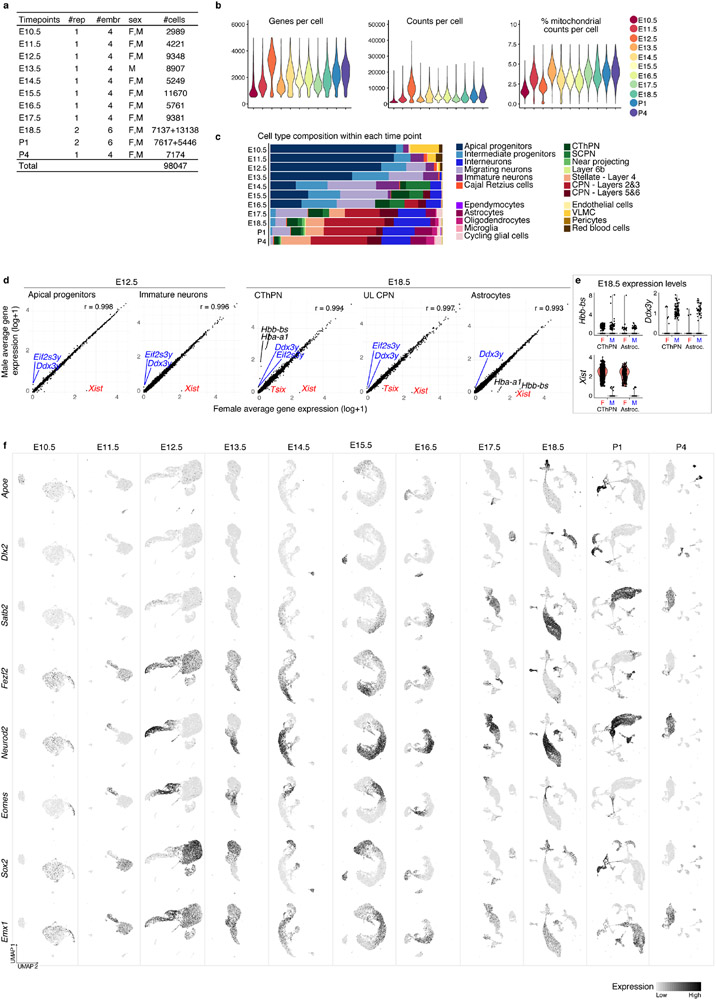
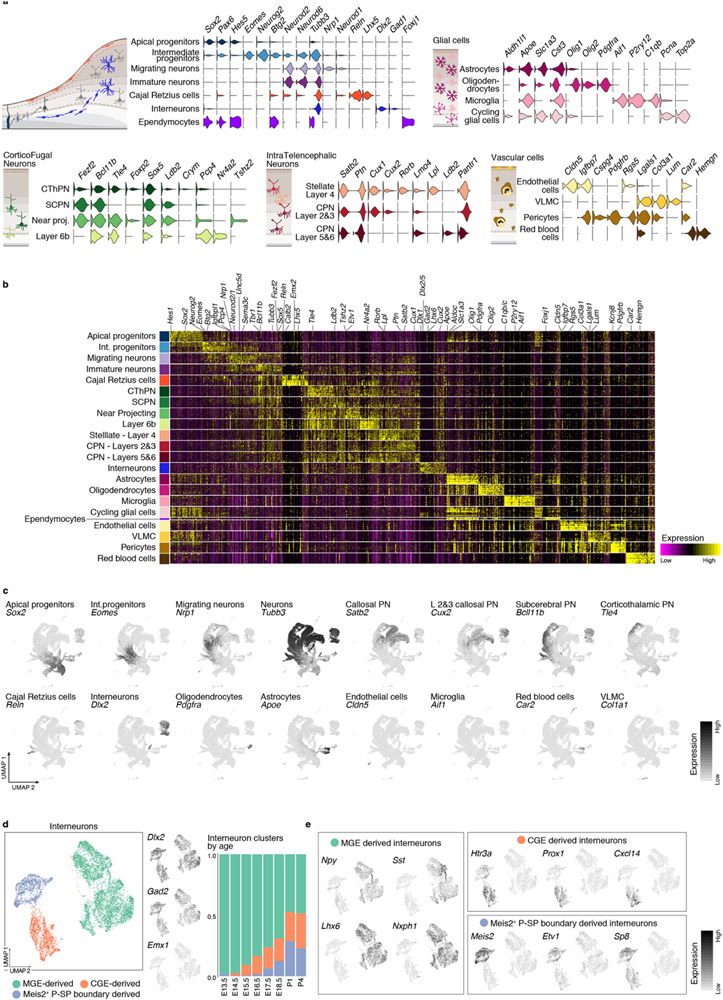
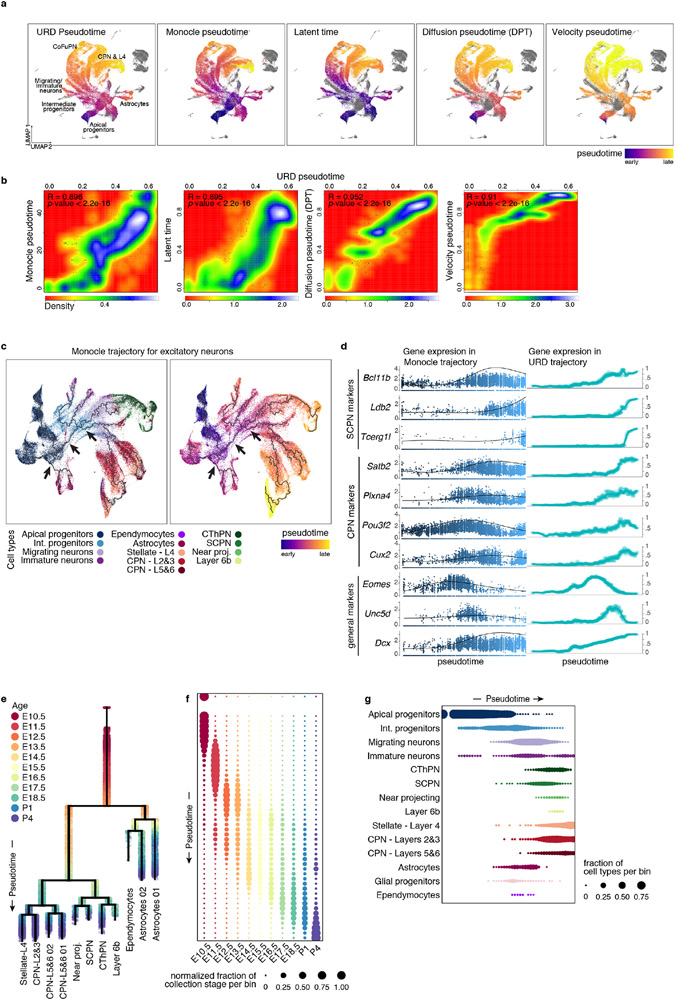
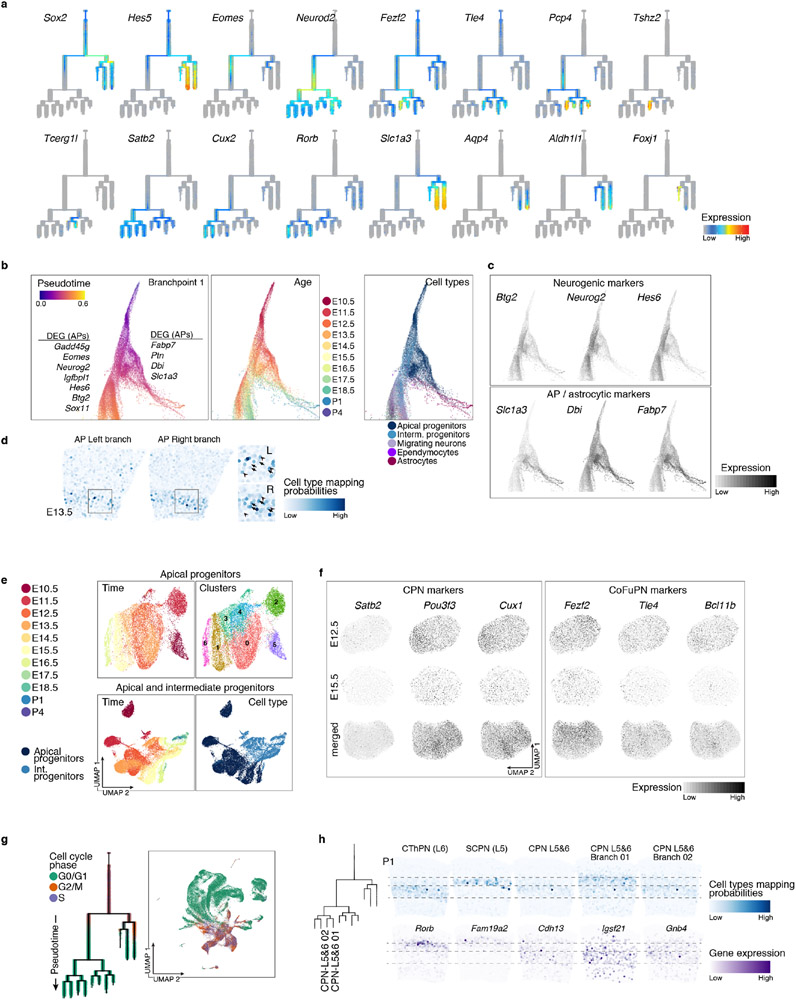
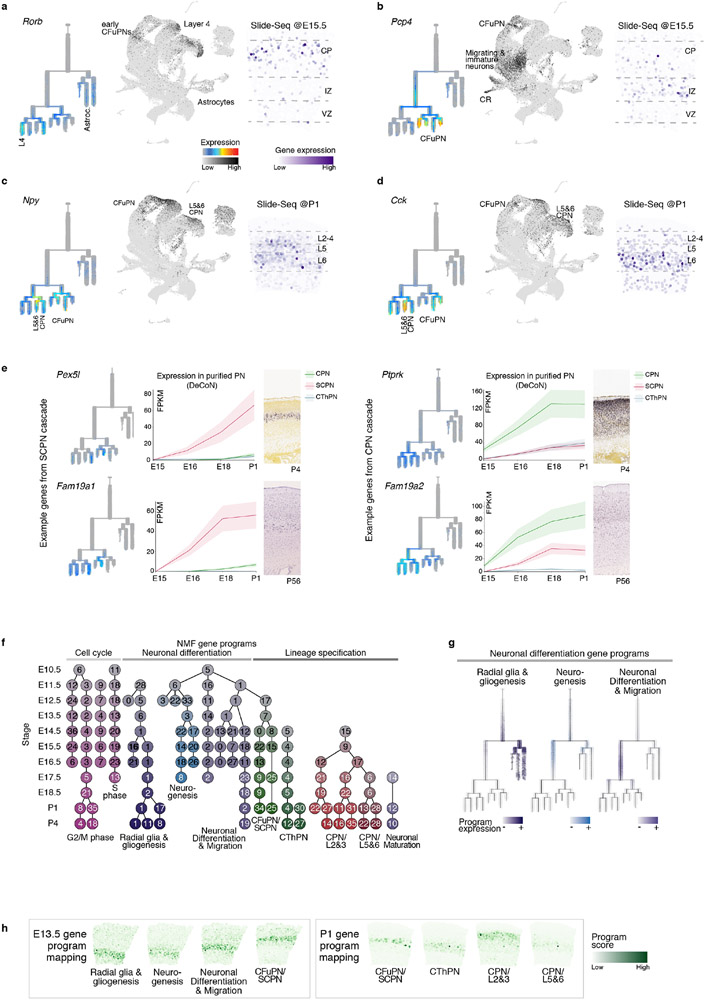
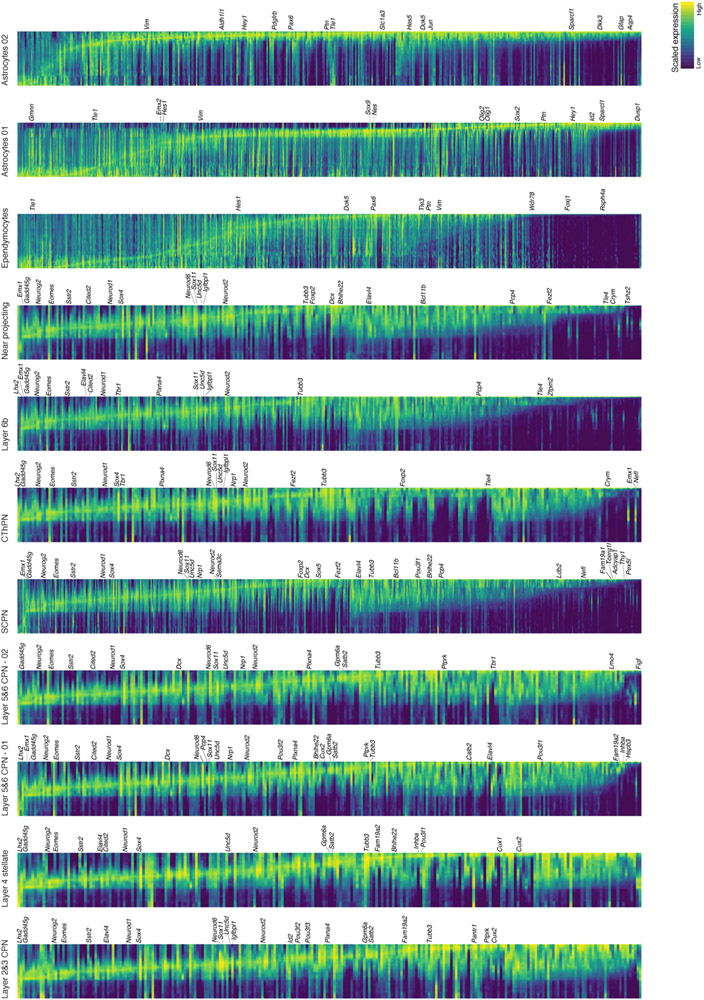
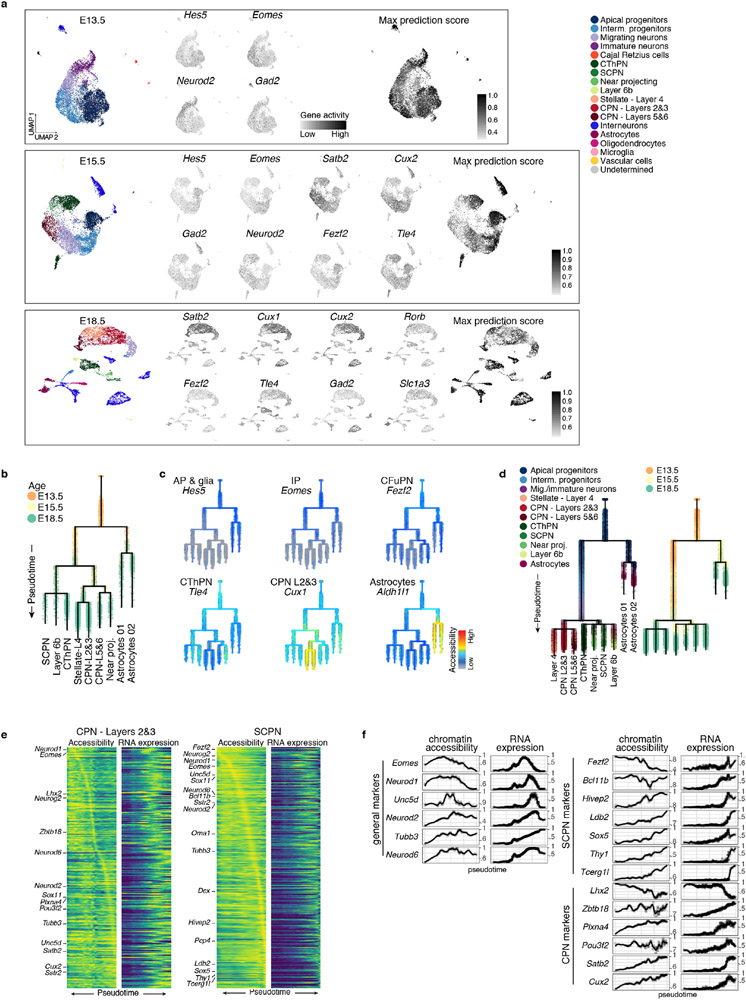
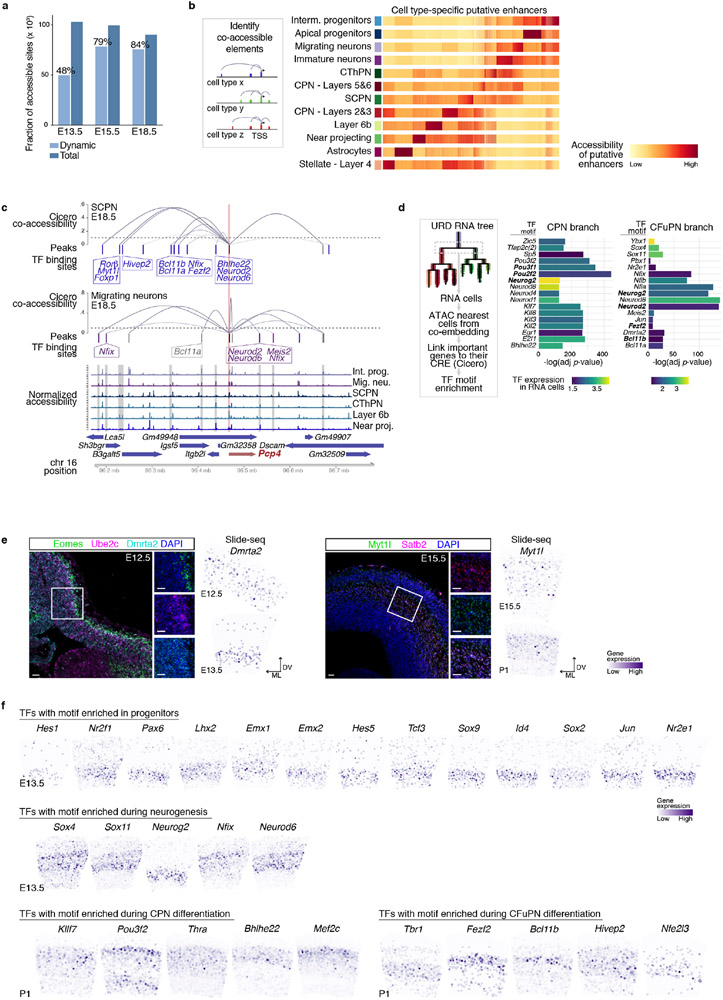
The mammalian cerebral cortex has an unparalleled diversity of cell types, which are generated during development through a series of temporally orchestrated events that are under tight evolutionary constraint and are critical for proper cortical assembly and function1,2. However, the molecular logic that governs the establishment and organization of cortical cell types remains unknown, largely due to the large number of cell classes that undergo dynamic cell-state transitions over extended developmental timelines. Here we generate a comprehensive atlas of the developing mouse neocortex, using single-cell RNA sequencing and single-cell assay for transposase-accessible chromatin using sequencing. We sampled the neocortex every day throughout embryonic corticogenesis and at early postnatal ages, and complemented the sequencing data with a spatial transcriptomics time course. We computationally reconstruct developmental trajectories across the diversity of cortical cell classes, and infer their spatial organization and the gene regulatory programs that accompany their lineage bifurcation decisions and differentiation trajectories. Finally, we demonstrate how this developmental map pinpoints the origin of lineage-specific developmental abnormalities that are linked to aberrant corticogenesis in mutant mice. The data provide a global picture of the regulatory mechanisms that govern cellular diversification in the neocortex.
© 2021. The Author(s), under exclusive licence to Springer Nature Limited.
Figures



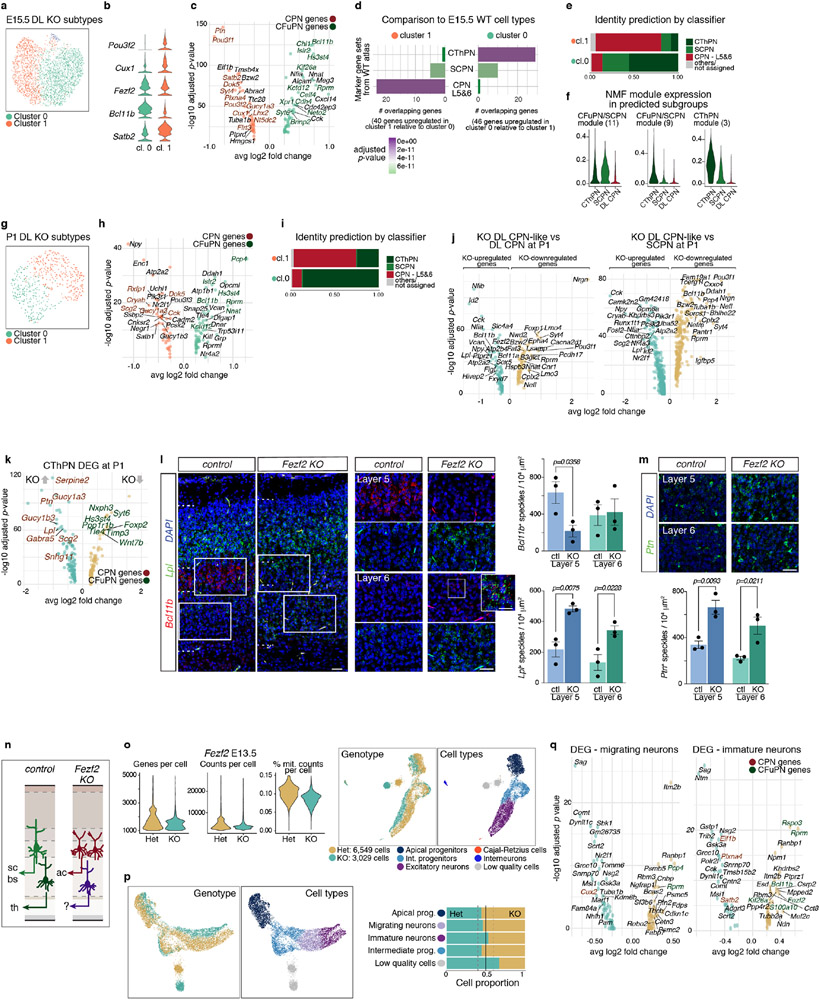
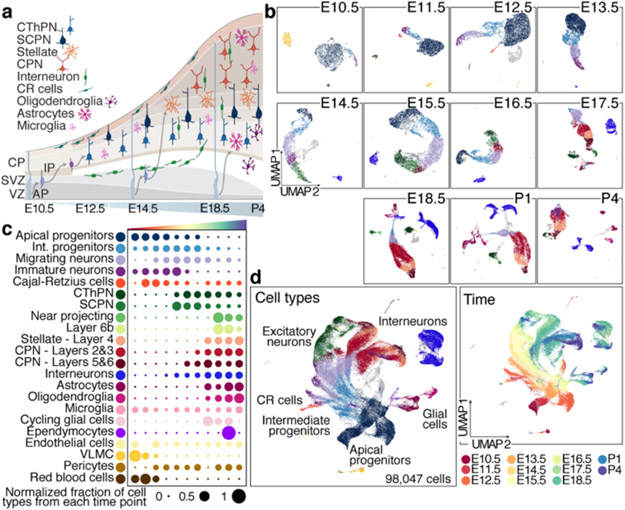
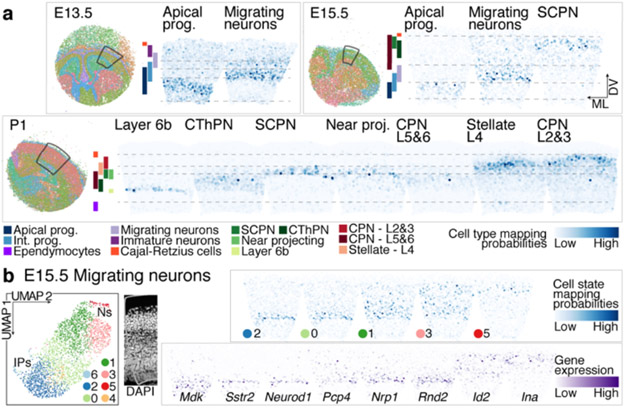
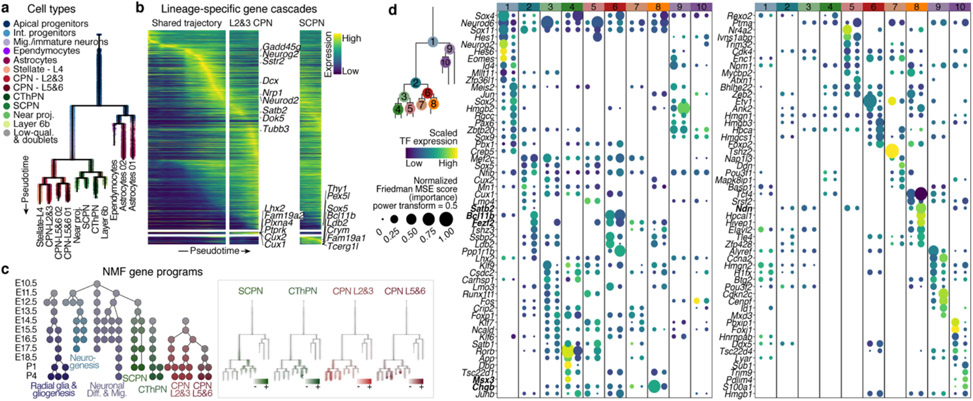

References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
