

A systematic CRISPR screen defines mutational mechanisms underpinning signatures caused by replication errors and endogenous DNA damage
- PMID: 34164627
- PMCID: PMC7611045
- DOI: 10.1038/s43018-021-00200-0
A systematic CRISPR screen defines mutational mechanisms underpinning signatures caused by replication errors and endogenous DNA damage
Abstract
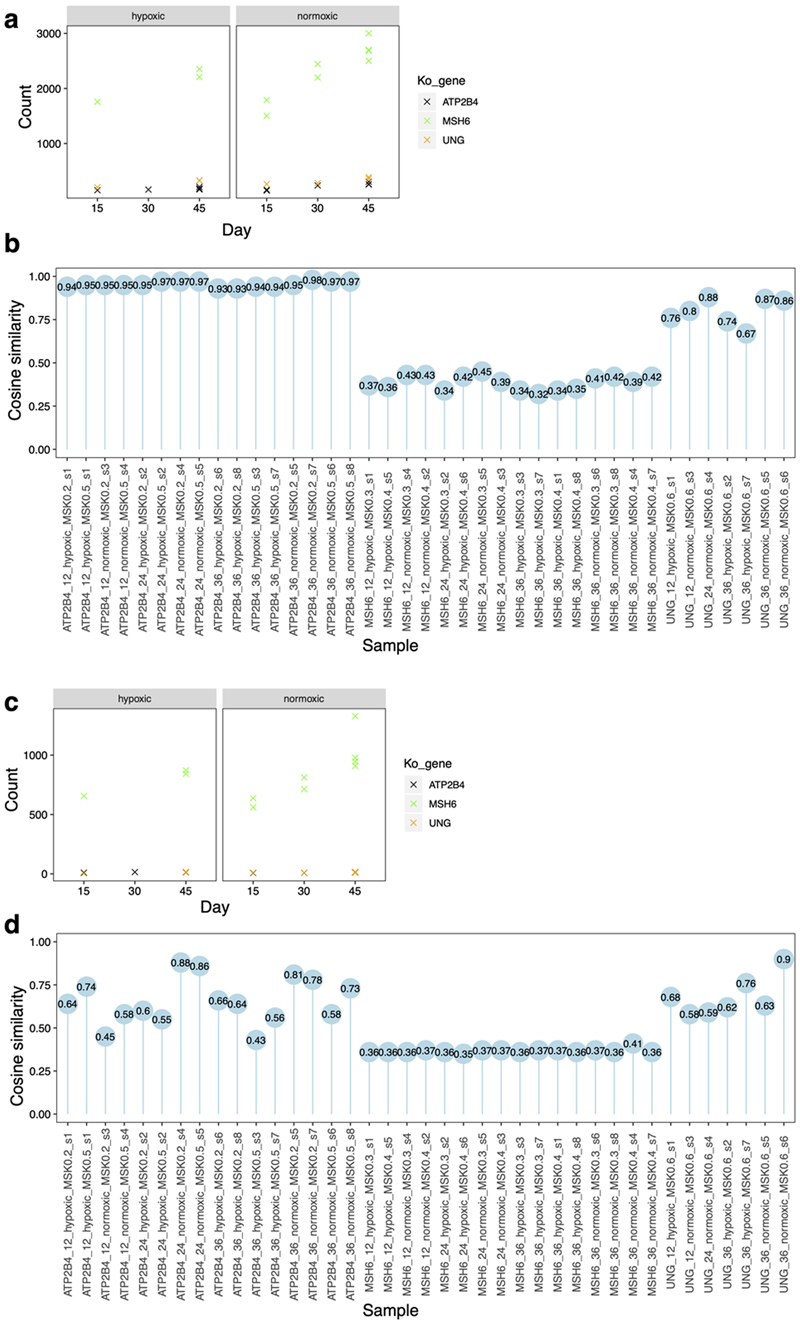
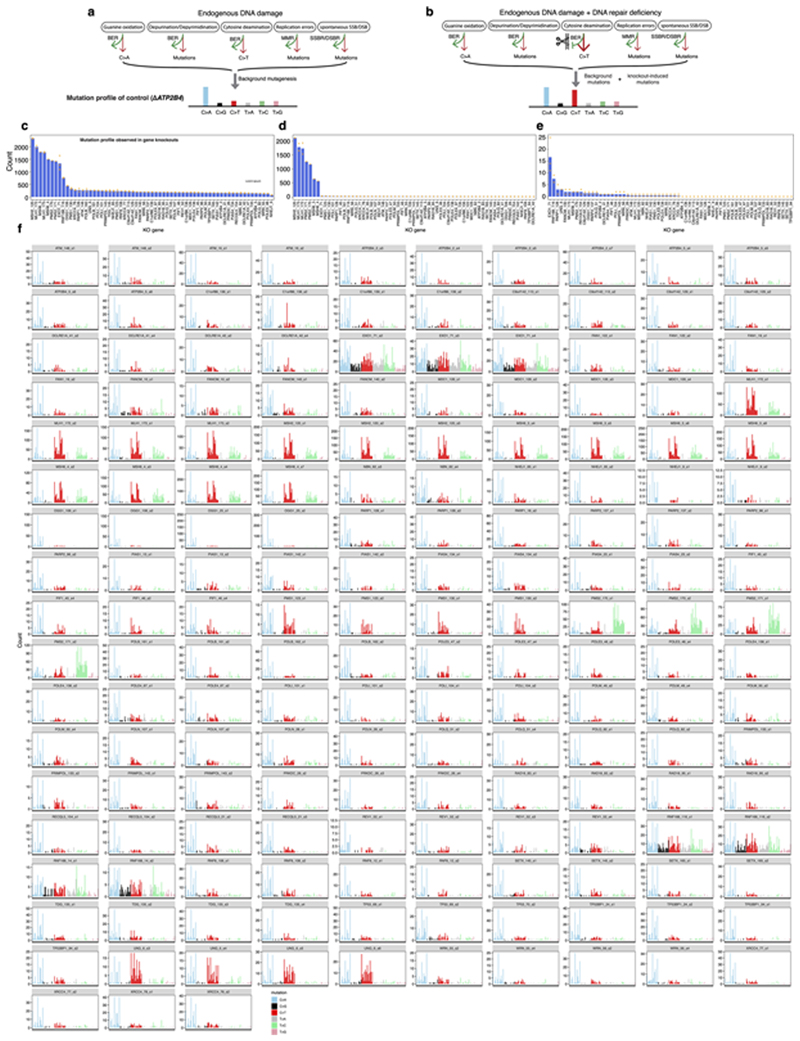
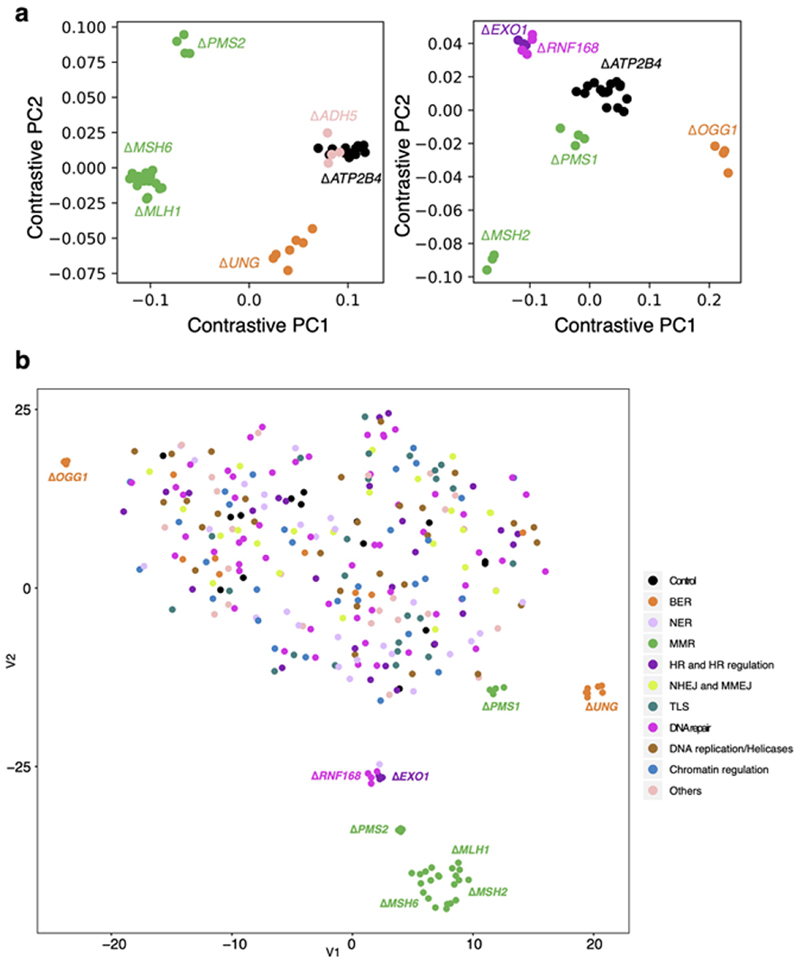
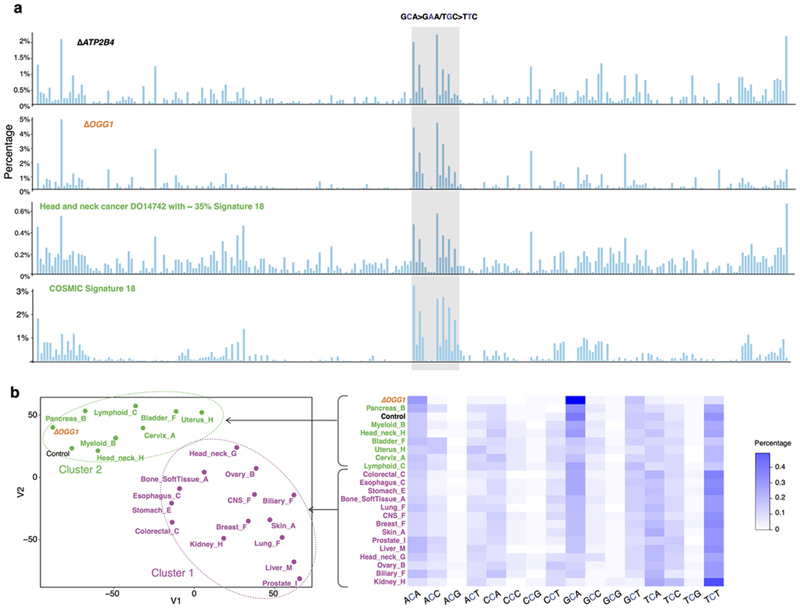
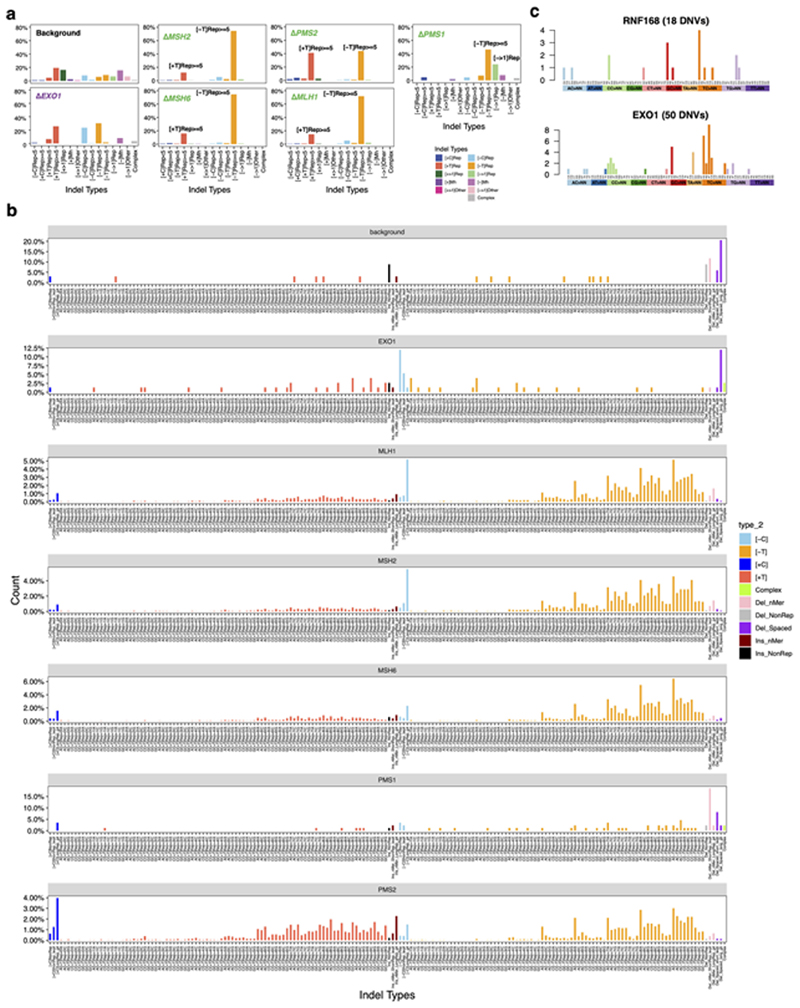
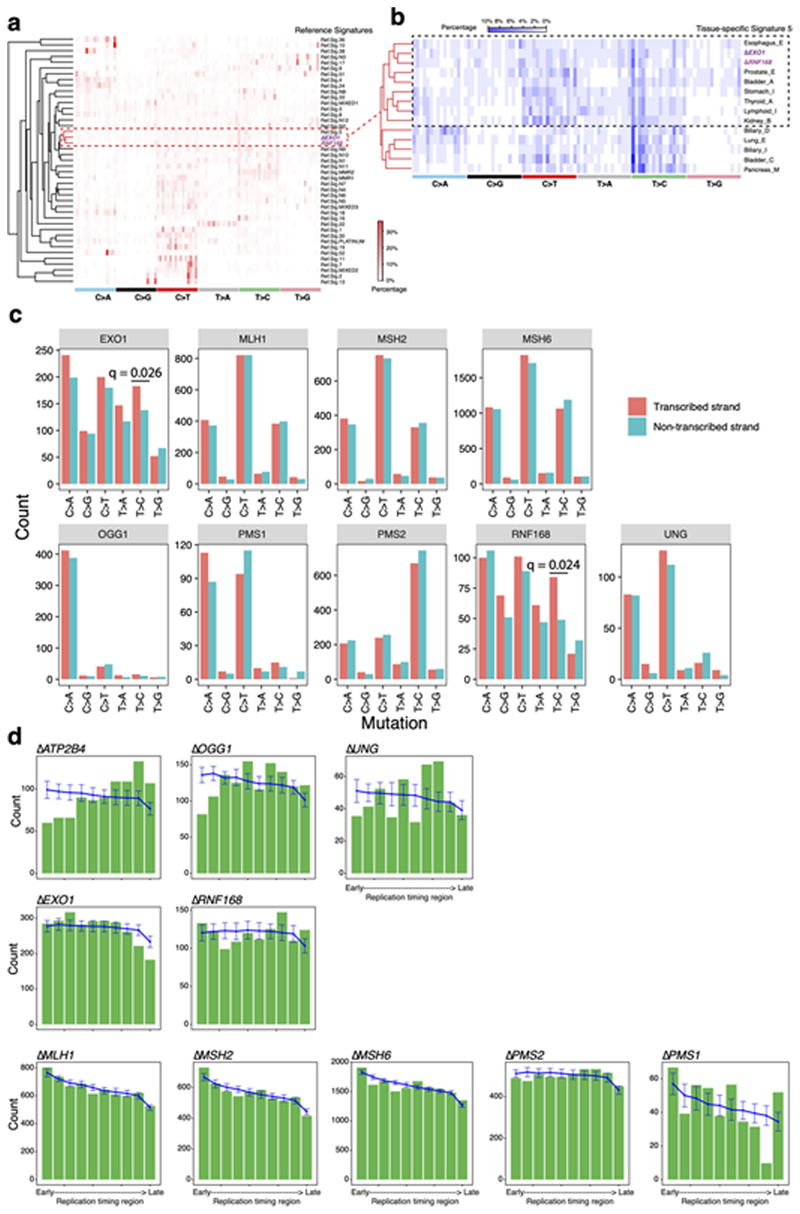
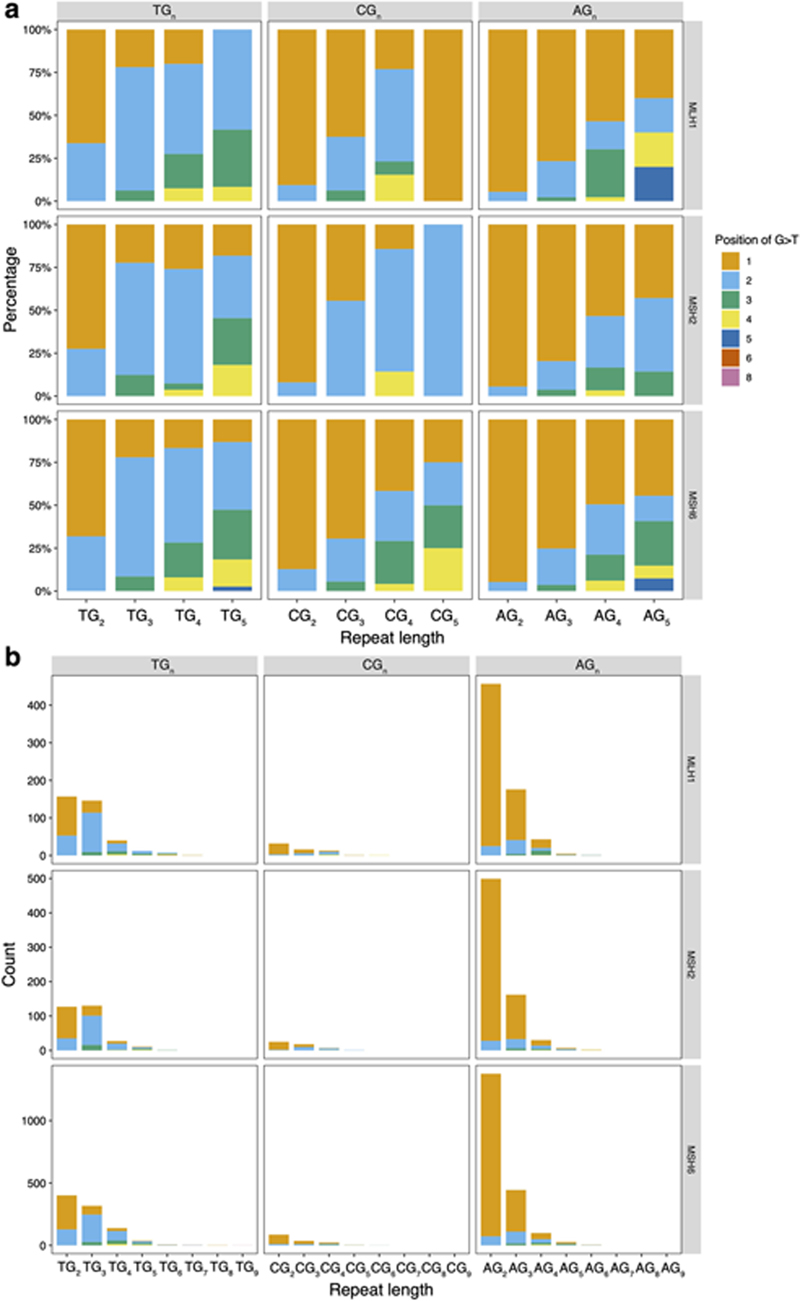
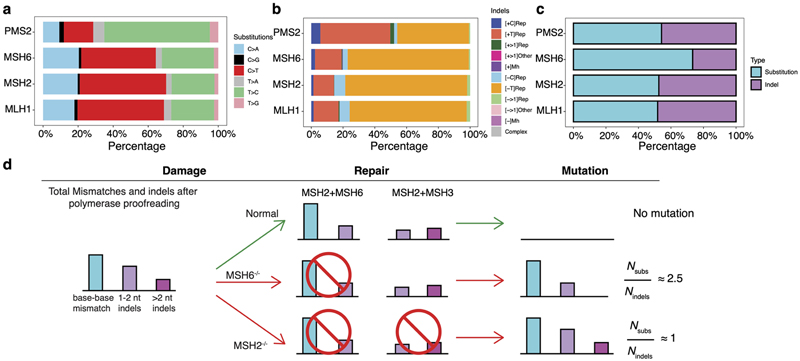
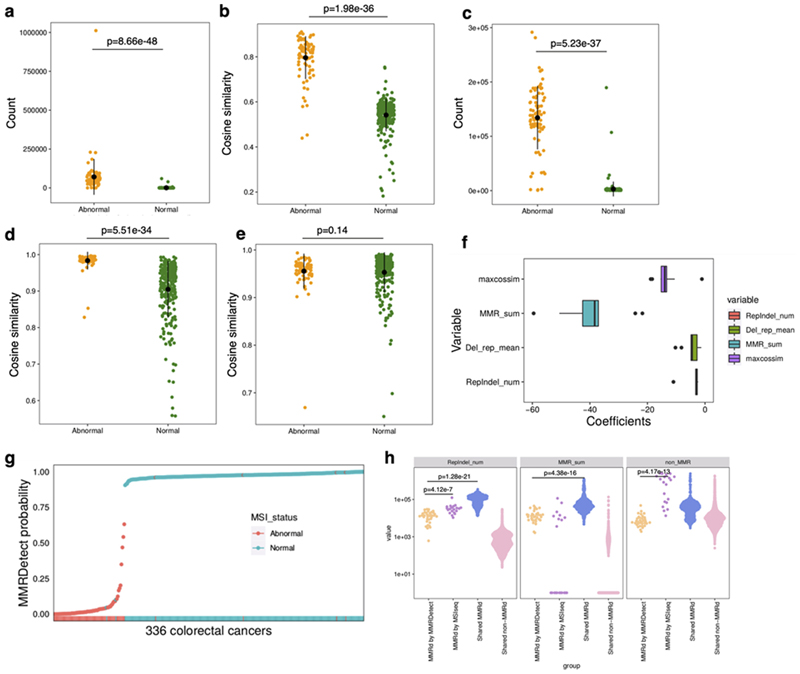
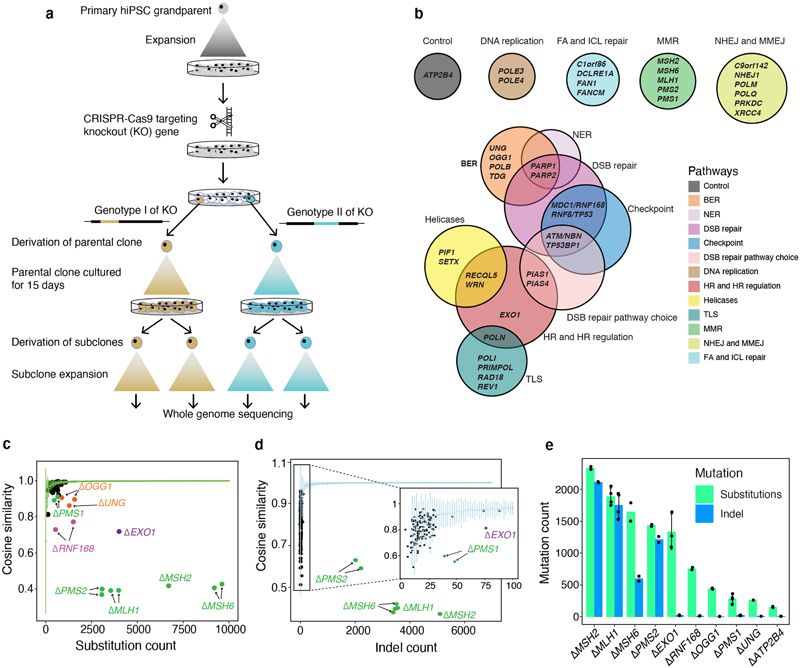
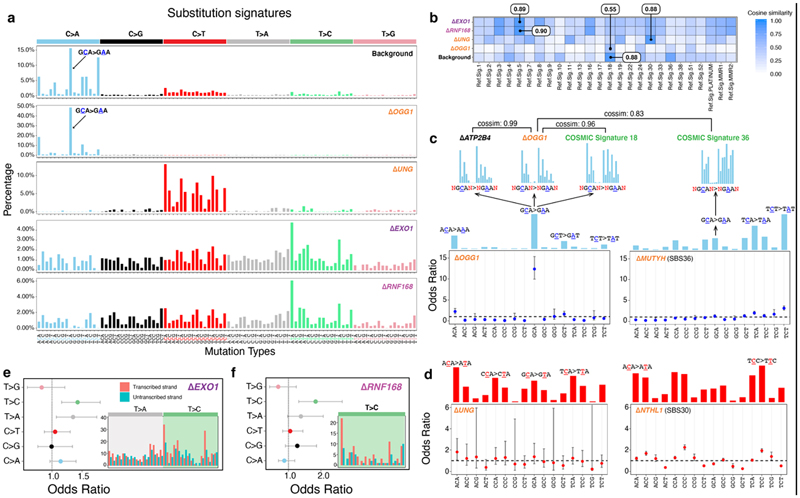
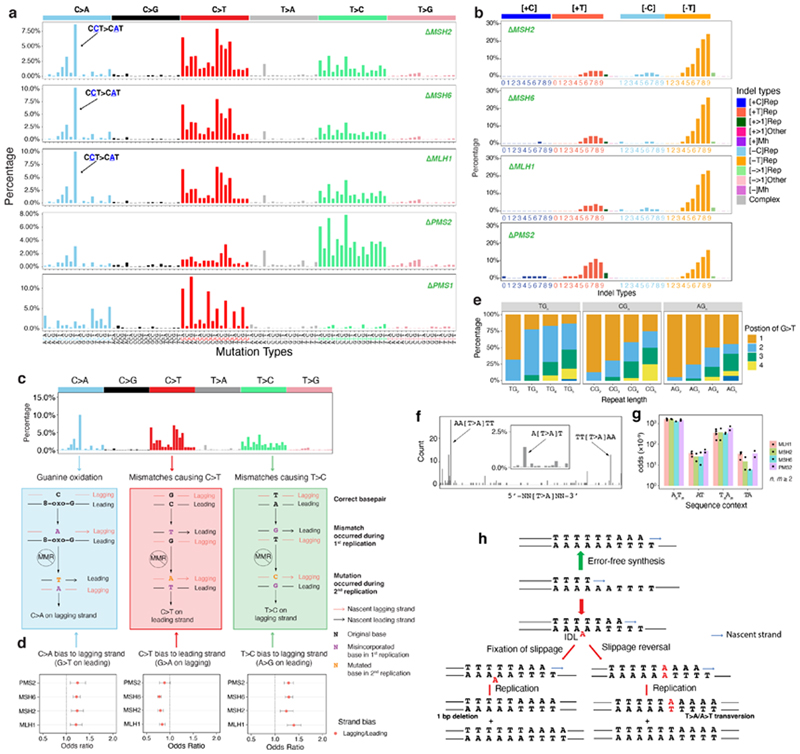
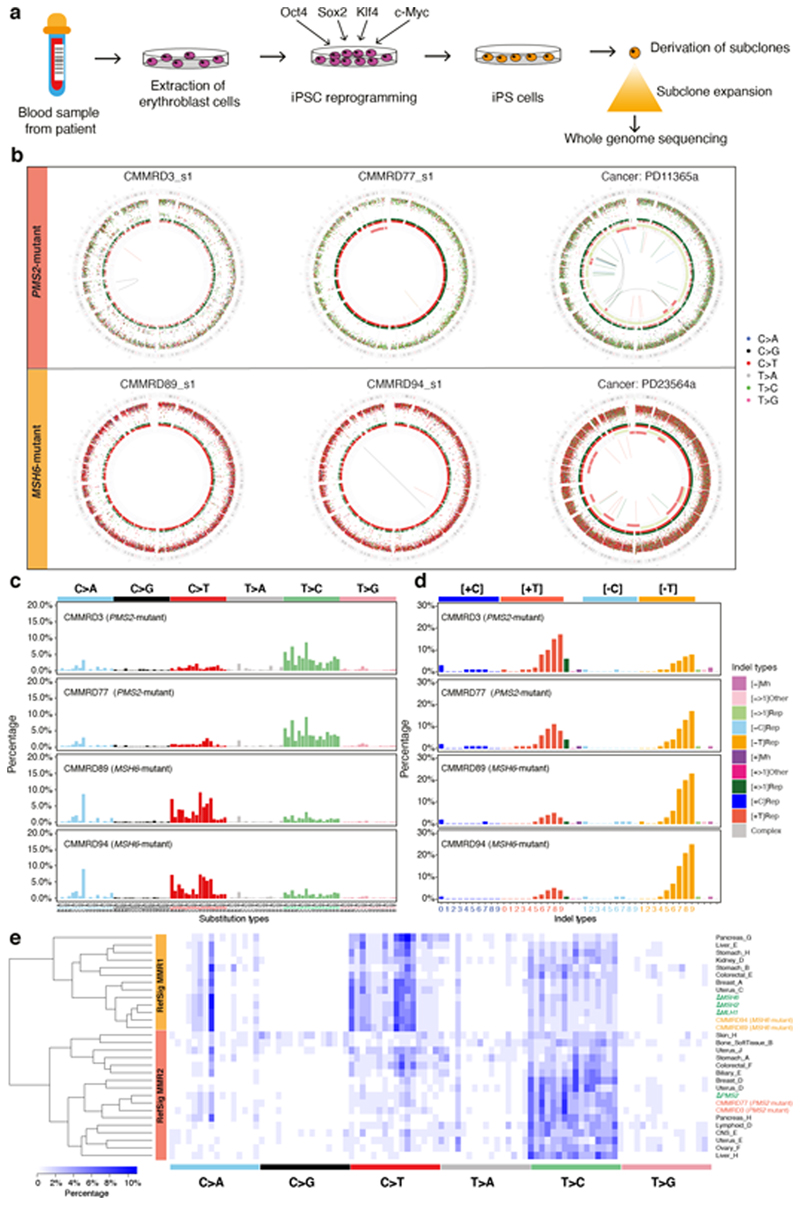
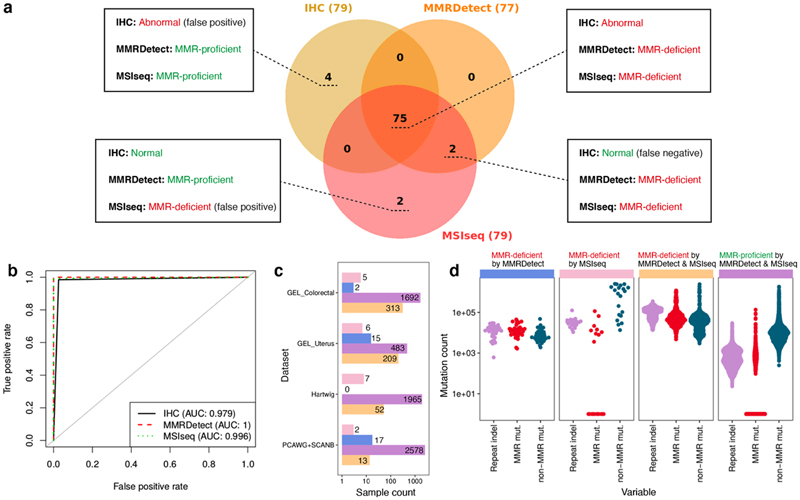
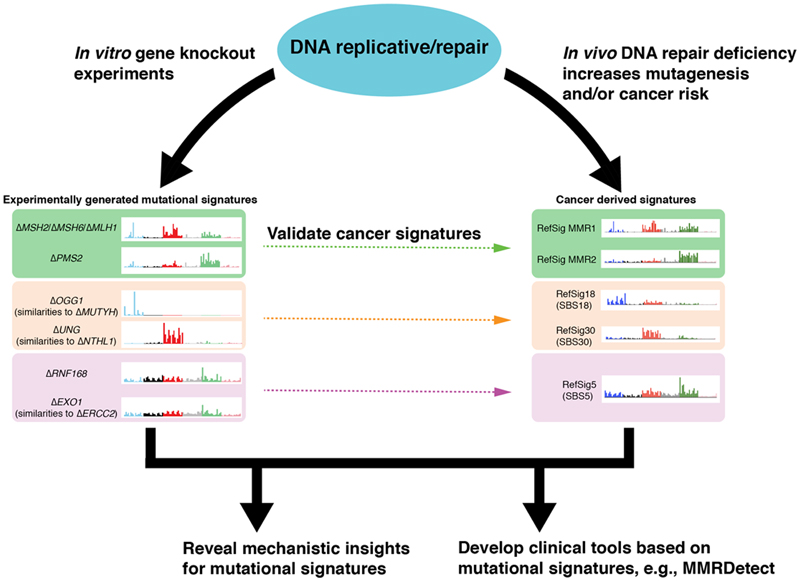
Mutational signatures are imprints of pathophysiological processes arising through tumorigenesis. We generated isogenic CRISPR-Cas9 knockouts (Δ) of 43 genes in human induced pluripotent stem cells, cultured them in the absence of added DNA damage, and performed whole-genome sequencing of 173 subclones. ΔOGG1, ΔUNG, ΔEXO1, ΔRNF168, ΔMLH1, ΔMSH2, ΔMSH6, ΔPMS1, and ΔPMS2 produced marked mutational signatures indicative of being critical mitigators of endogenous DNA modifications. Detailed analyses revealed mutational mechanistic insights, including how 8-oxo-dG elimination is sequence-context-specific while uracil clearance is sequence-context-independent. Mismatch repair (MMR) deficiency signatures are engendered by oxidative damage (C>A transversions), differential misincorporation by replicative polymerases (T>C and C>T transitions), and we propose a 'reverse template slippage' model for T>A transversions. ΔMLH1, ΔMSH6, and ΔMSH2 signatures were similar to each other but distinct from ΔPMS2. Finally, we developed a classifier, MMRDetect, where application to 7,695 WGS cancers showed enhanced detection of MMR-deficient tumors, with implications for responsiveness to immunotherapies.
Keywords: CRISPR-Cas9 systems; Genomic instability; cancer; cancer genomics.
Conflict of interest statement
Competing Interests Statement SNZ holds patents on clinical algorithms of mutational signatures and during the completion of this project, served advisory roles for Astra Zeneca, Artios Pharma Ltd and Scottish Genome Project.
Figures

References
Publication types
MeSH terms
Supplementary concepts
Grants and funding
- A23916/CRUK_/Cancer Research UK/United Kingdom
- 23433/CRUK_/Cancer Research UK/United Kingdom
- 23916/CRUK_/Cancer Research UK/United Kingdom
- MR/R015724/1/MRC_/Medical Research Council/United Kingdom
- MC_PC_14089/MRC_/Medical Research Council/United Kingdom
- MR/M009203/1/MRC_/Medical Research Council/United Kingdom
- C309/A25144/CRUK_/Cancer Research UK/United Kingdom
- A23433/CRUK_/Cancer Research UK/United Kingdom
- 100183/WT_/Wellcome Trust/United Kingdom
- A22932/CRUK_/Cancer Research UK/United Kingdom
- MC_EX_MR/M009203/1/MRC_/Medical Research Council/United Kingdom
- 101126/WT_/Wellcome Trust/United Kingdom
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
