

Cerebral Cavernous Malformation: From Mechanism to Therapy
- PMID: 34166073
- PMCID: PMC8922476
- DOI: 10.1161/CIRCRESAHA.121.318174
Cerebral Cavernous Malformation: From Mechanism to Therapy
Erratum in
-
Correction to: Cerebral Cavernous Malformation: From Mechanism to Therapy.Circ Res. 2021 Aug 6;129(4):e101. doi: 10.1161/RES.0000000000000496. Epub 2021 Aug 5. Circ Res. 2021. PMID: 34351801 No abstract available.
Abstract
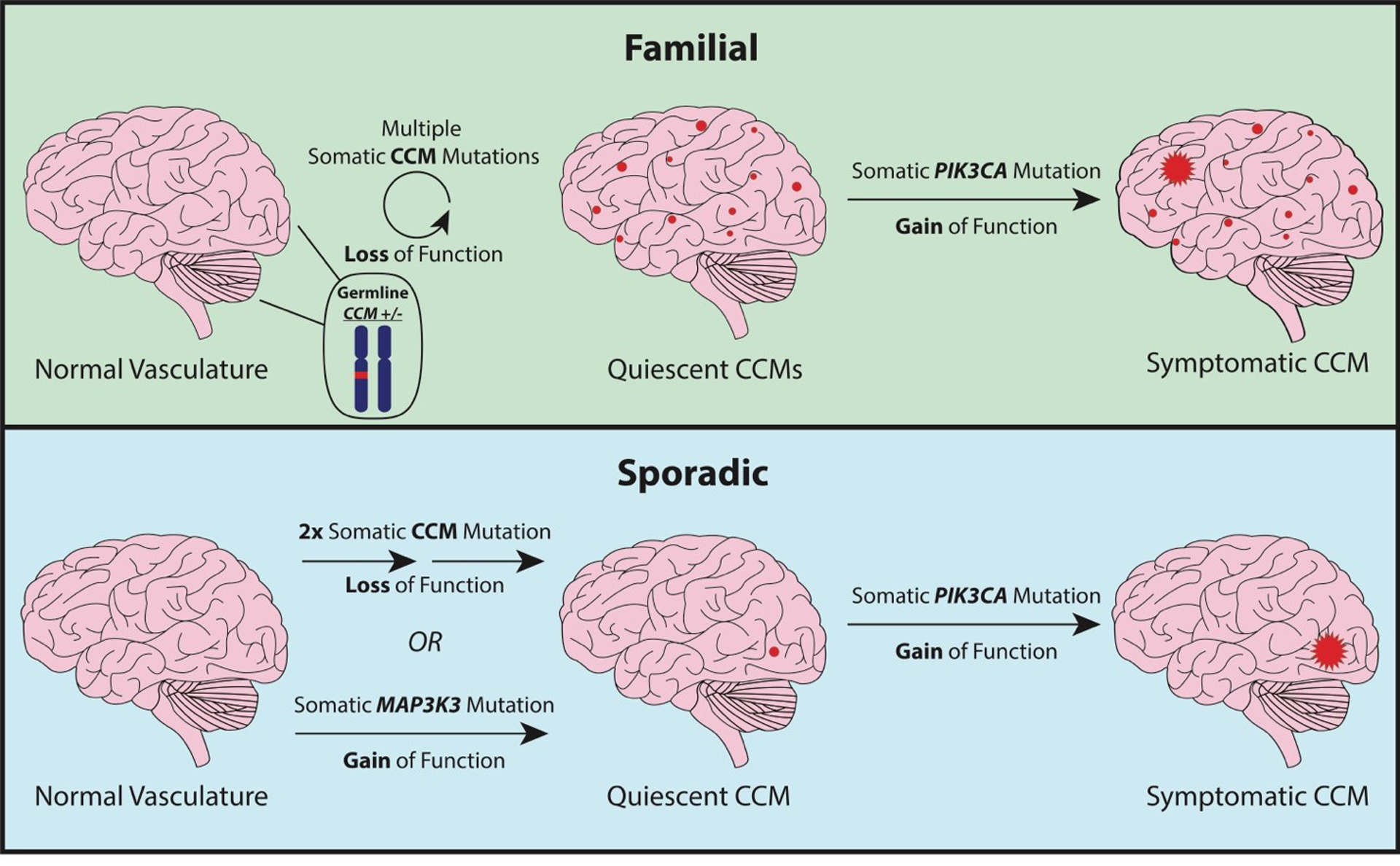
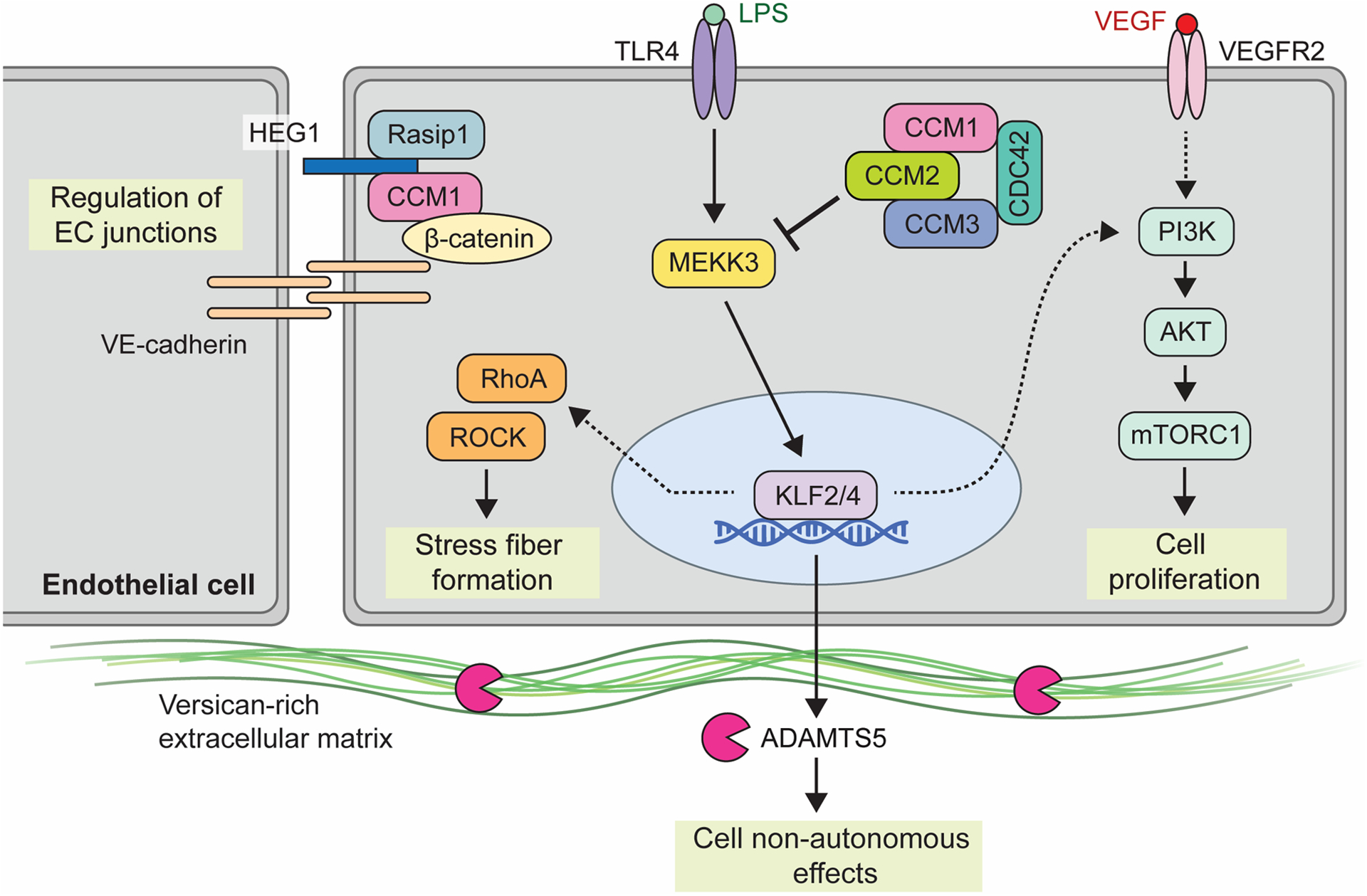
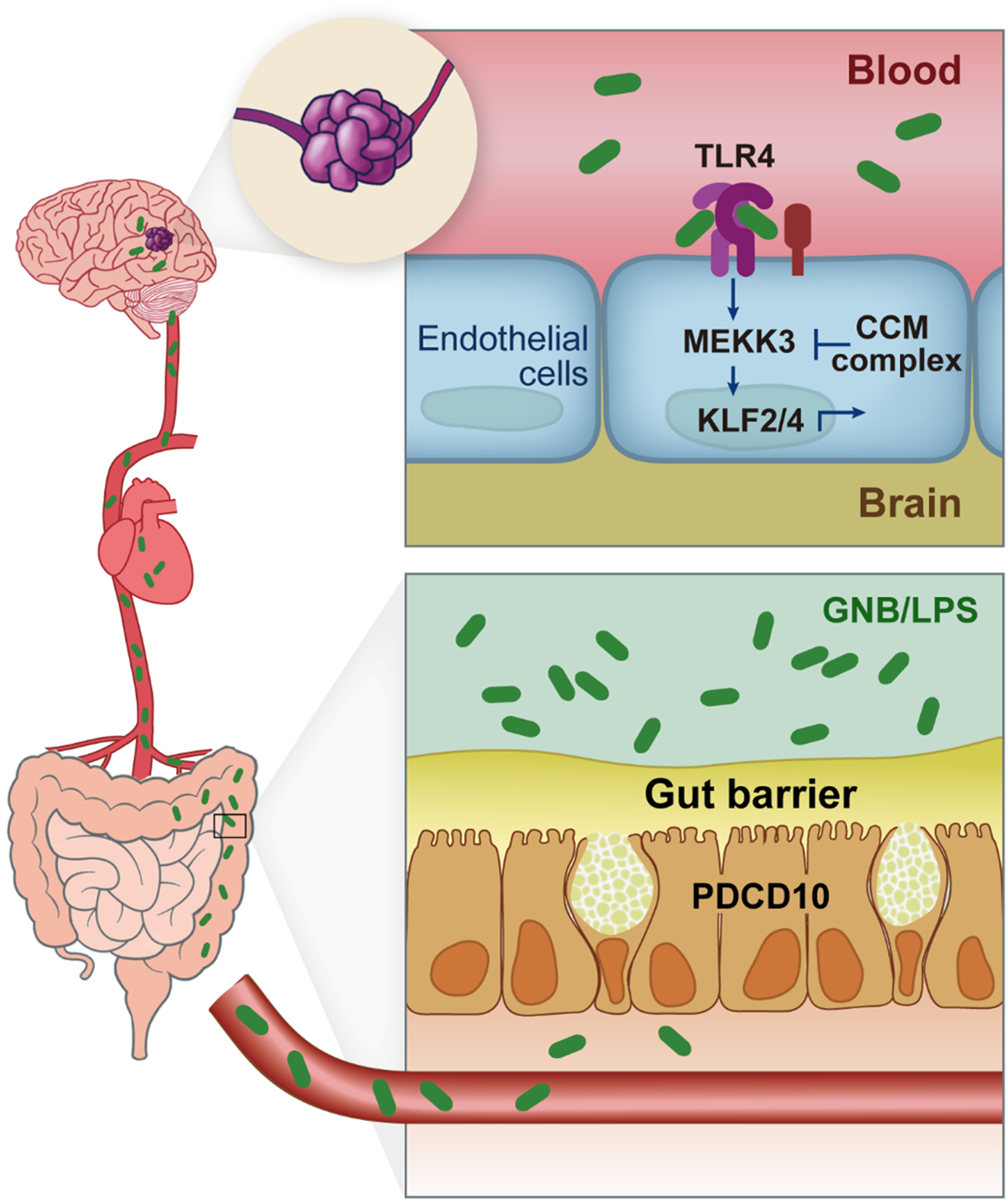
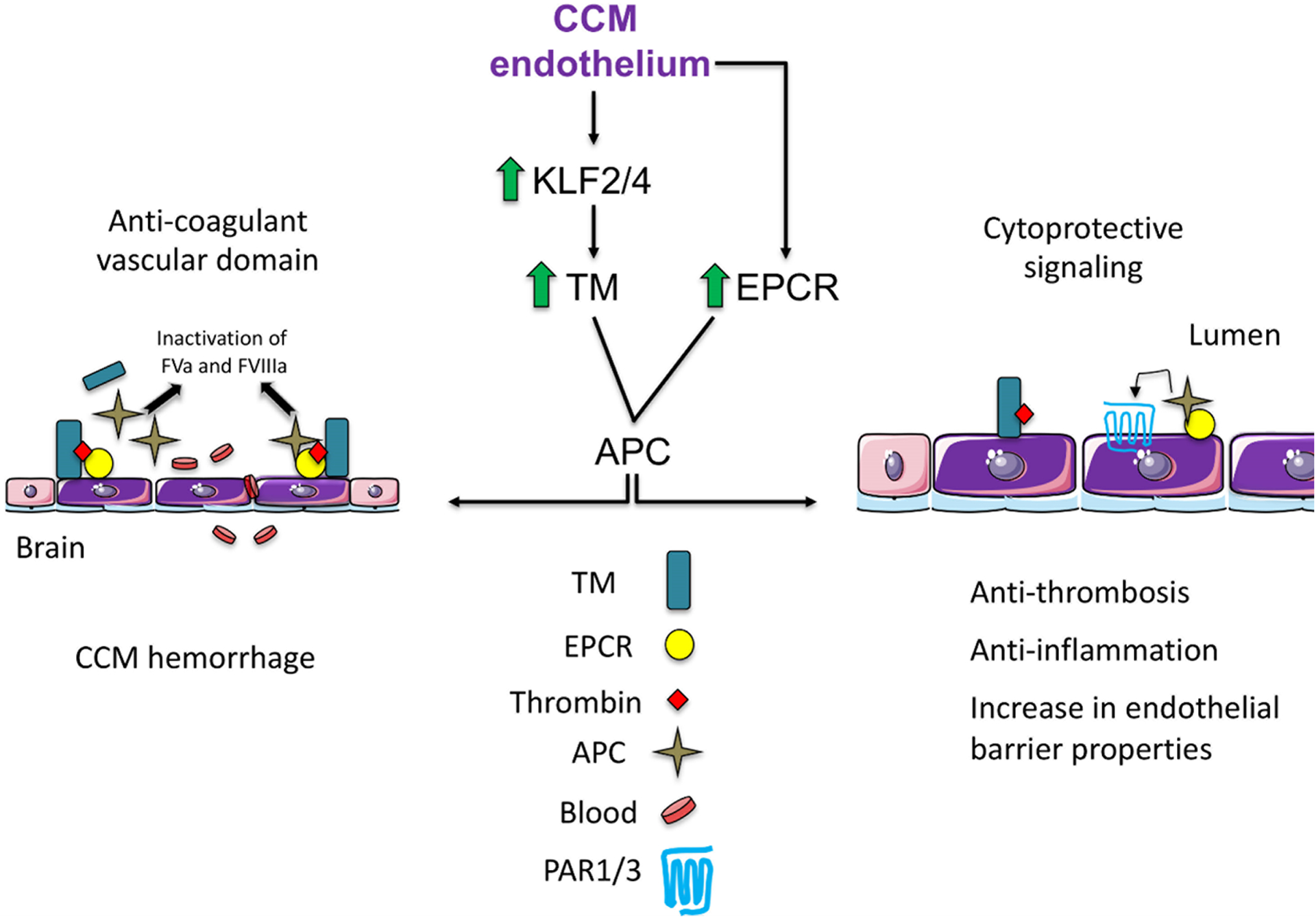
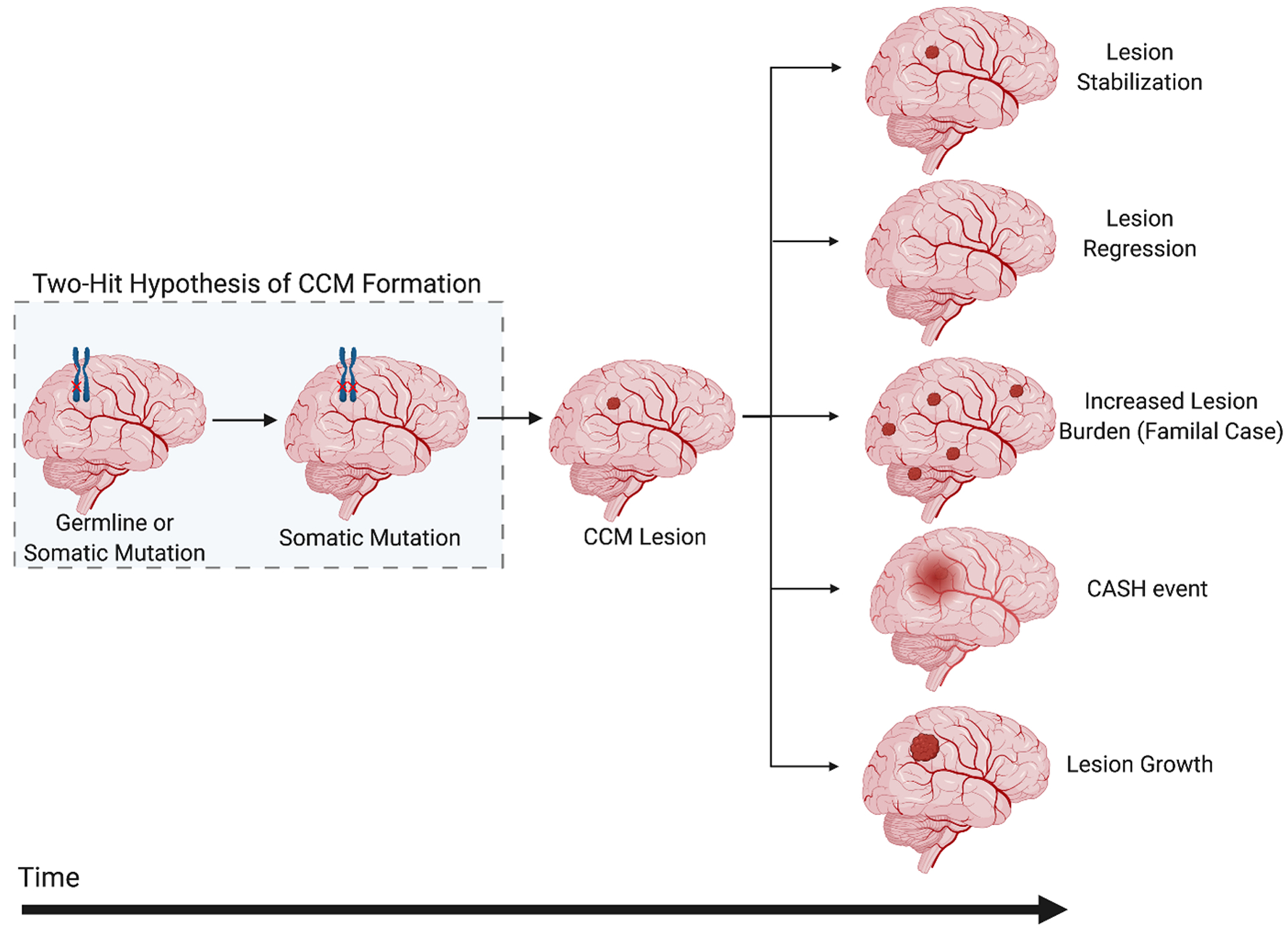
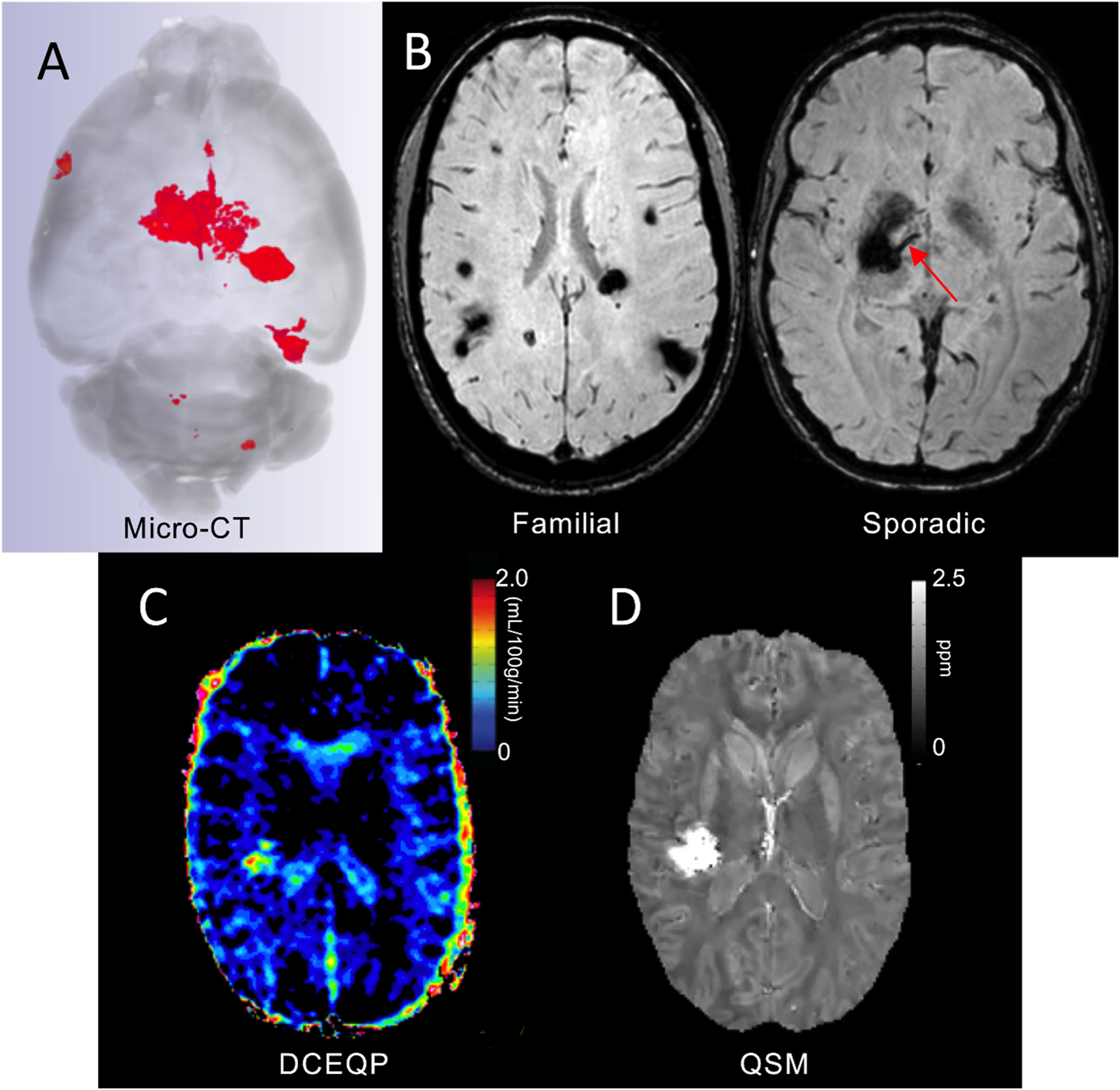
Cerebral cavernous malformations are acquired vascular anomalies that constitute a common cause of central nervous system hemorrhage and stroke. The past 2 decades have seen a remarkable increase in our understanding of the pathogenesis of this vascular disease. This new knowledge spans genetic causes of sporadic and familial forms of the disease, molecular signaling changes in vascular endothelial cells that underlie the disease, unexpectedly strong environmental effects on disease pathogenesis, and drivers of disease end points such as hemorrhage. These novel insights are the integrated product of human clinical studies, human genetic studies, studies in mouse and zebrafish genetic models, and basic molecular and cellular studies. This review addresses the genetic and molecular underpinnings of cerebral cavernous malformation disease, the mechanisms that lead to lesion hemorrhage, and emerging biomarkers and therapies for clinical treatment of cerebral cavernous malformation disease. It may also serve as an example for how focused basic and clinical investigation and emerging technologies can rapidly unravel a complex disease mechanism.
Keywords: central nervous system; endothelial cells; hemorrhage; stroke; vascular malformations.
Figures
References
-
- Bicknell JM, Carlow TJ, Kornfeld M, Stovring J and Turner P. Familial cavernous angiomas. Arch Neurol. 1978;35:746–9. - PubMed
-
- Kidd HA and Cumings JN. Cerebral angiomata in an Icelandic family. Lancet. 1947;1:747. - PubMed
-
- Laberge-le Couteulx S, Jung HH, Labauge P, Houtteville JP, Lescoat C, Cecillon M, Marechal E, Joutel A, Bach JF and Tournier-Lasserve E. Truncating mutations in CCM1, encoding KRIT1, cause hereditary cavernous angiomas. Nat Genet. 1999;23:189–93. - PubMed
-
- Sahoo T, Johnson EW, Thomas JW, Kuehl PM, Jones TL, Dokken CG, Touchman JW, Gallione CJ, Lee-Lin SQ, Kosofsky B, Kurth JH, Louis DN, Mettler G, Morrison L, Gil-Nagel A, Rich SS, Zabramski JM, Boguski MS, Green ED and Marchuk DA. Mutations in the gene encoding KRIT1, a Krev-1/rap1a binding protein, cause cerebral cavernous malformations (CCM1). Hum Mol Genet. 1999;8:2325–33. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
