

ATR Inhibition Induces CDK1-SPOP Signaling and Enhances Anti-PD-L1 Cytotoxicity in Prostate Cancer
- PMID: 34168048
- PMCID: PMC8456453
- DOI: 10.1158/1078-0432.CCR-21-1010
ATR Inhibition Induces CDK1-SPOP Signaling and Enhances Anti-PD-L1 Cytotoxicity in Prostate Cancer
Abstract
Purpose: Despite significant benefit for other cancer subtypes, immune checkpoint blockade (ICB) therapy has not yet been shown to significantly improve outcomes for men with castration-resistant prostate cancer (CRPC). Prior data have shown that DNA damage response (DDR) deficiency, via genetic alteration and/or pharmacologic induction using DDR inhibitors (DDRi), may improve ICB response in solid tumors in part due to induction of mitotic catastrophe and innate immune activation. Discerning the underlying mechanisms of this DDRi-ICB interaction in a prostate cancer-specific manner is vital to guide novel clinical trials and provide durable clinical responses for men with CRPC.
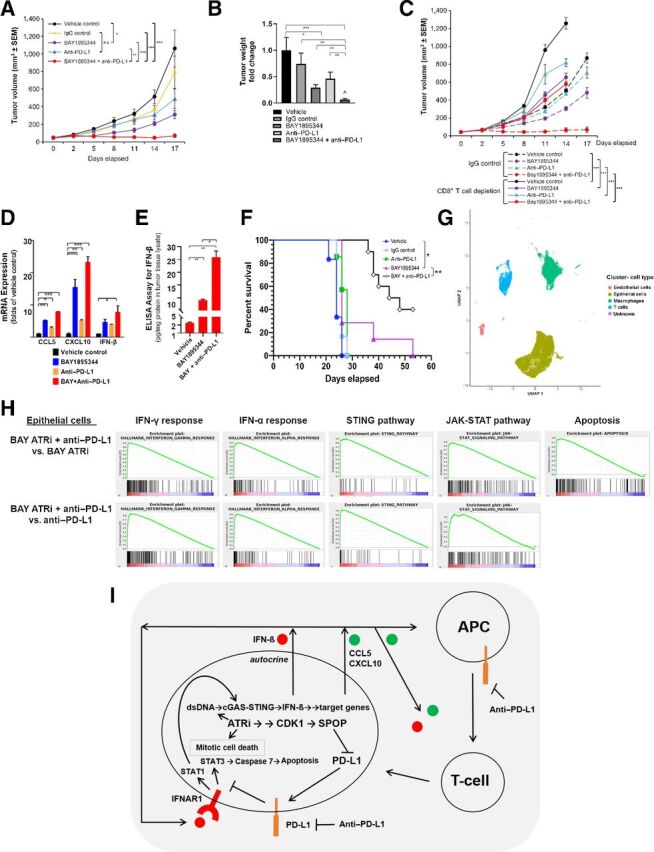
Experimental design: We treated prostate cancer cell lines with potent, specific inhibitors of ATR kinase, as well as with PARP inhibitor, olaparib. We performed analyses of cGAS-STING and DDR signaling in treated cells, and treated a syngeneic androgen-indifferent, prostate cancer model with combined ATR inhibition and anti-programmed death ligand 1 (anti-PD-L1), and performed single-cell RNA sequencing analysis in treated tumors.
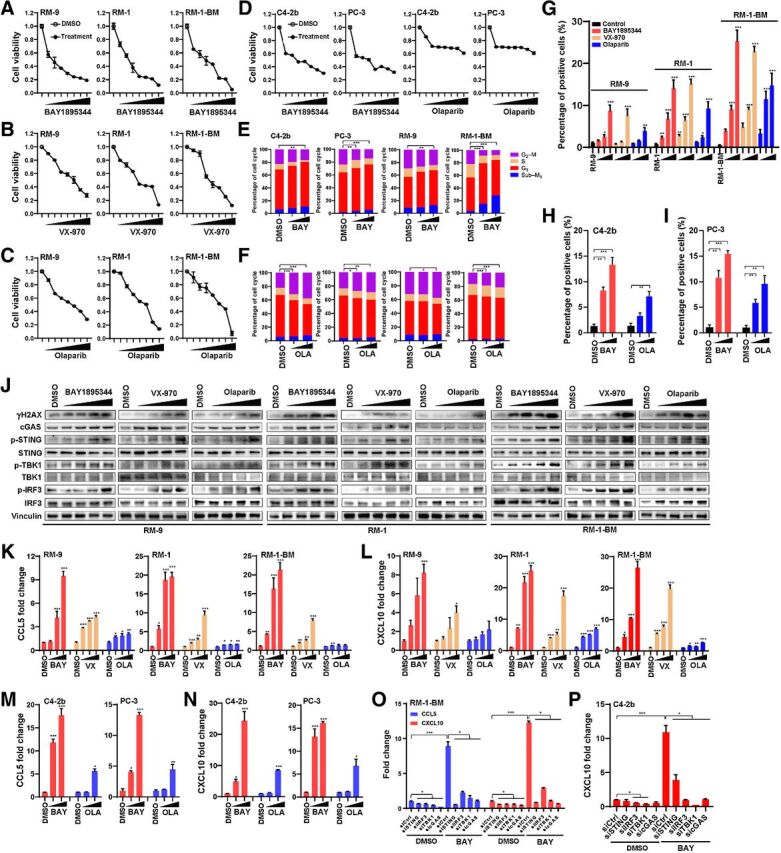
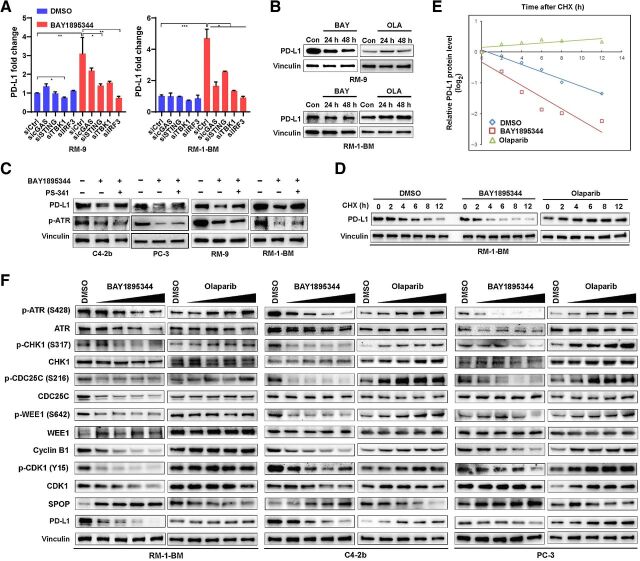
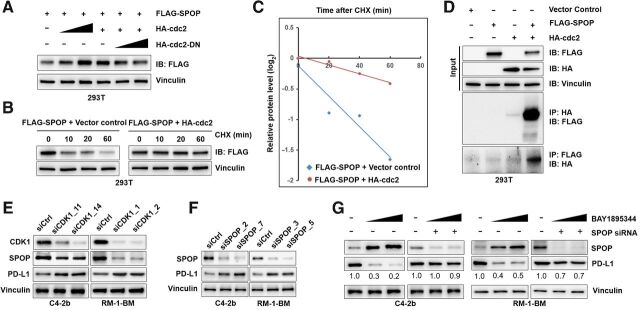
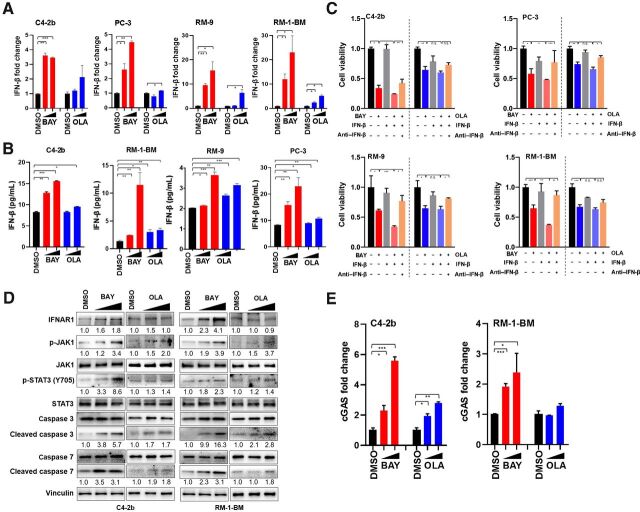
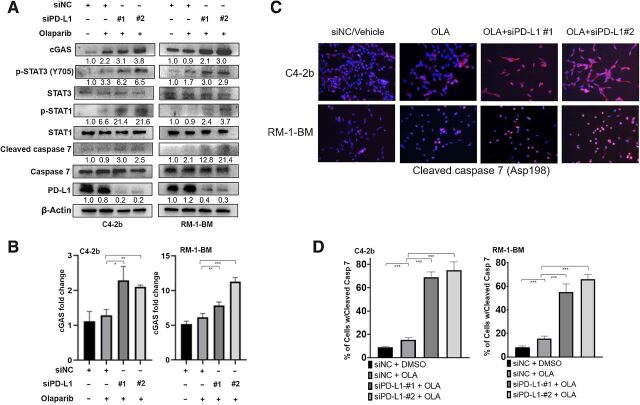
Results: ATR inhibitor (ATRi; BAY1895433) directly repressed ATR-CHK1 signaling, activated CDK1-SPOP axis, leading to destabilization of PD-L1 protein. These effects of ATRi are distinct from those of olaparib, and resulted in a cGAS-STING-initiated, IFN-β-mediated, autocrine, apoptotic response in CRPC. The combination of ATRi with anti-PD-L1 therapy resulted in robust innate immune activation and a synergistic, T-cell-dependent therapeutic response in our syngeneic mouse model.
Conclusions: This work provides a molecular mechanistic rationale for combining ATR-targeted agents with immune checkpoint blockade for patients with CRPC. Multiple early-phase clinical trials of this combination are underway.
©2021 The Authors; Published by the American Association for Cancer Research.
Figures
References
-
- Beer TM, Kwon ED, Drake CG, Fizazi K, Logothetis C, Gravis G, et al. Randomized, double-blind, phase III trial of ipilimumab versus placebo in asymptomatic or minimally symptomatic patients with metastatic chemotherapy-naive castration-resistant prostate cancer. J Clin Oncol 2017;35:40–7. - PubMed
-
- Robinson D, Van Allen EM, Wu YM, Schultz N, Lonigro RJ, Mosquera JM, et al. Integrative clinical genomics of advanced prostate cancer. Cell 2015;162:454. - PubMed
-
- Subudhi SK, Vence L, Zhao H, Blando J, Yadav SS, Xiong Q, et al. Neoantigen responses, immune correlates, and favorable outcomes after ipilimumab treatment of patients with prostate cancer. Sci Transl Med 2020;12:eaaz3577. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
