

The architecture of the SARS-CoV-2 RNA genome inside virion
- PMID: 34168138
- PMCID: PMC8225788
- DOI: 10.1038/s41467-021-22785-x
The architecture of the SARS-CoV-2 RNA genome inside virion
Abstract
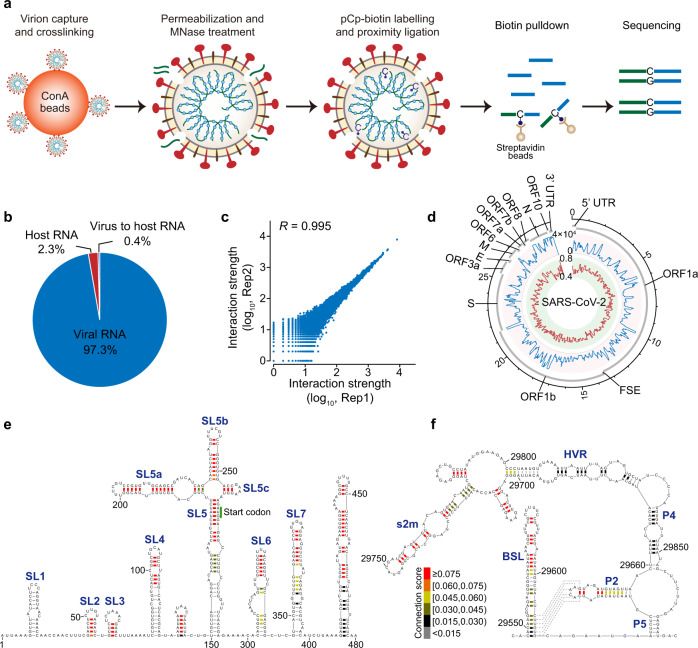
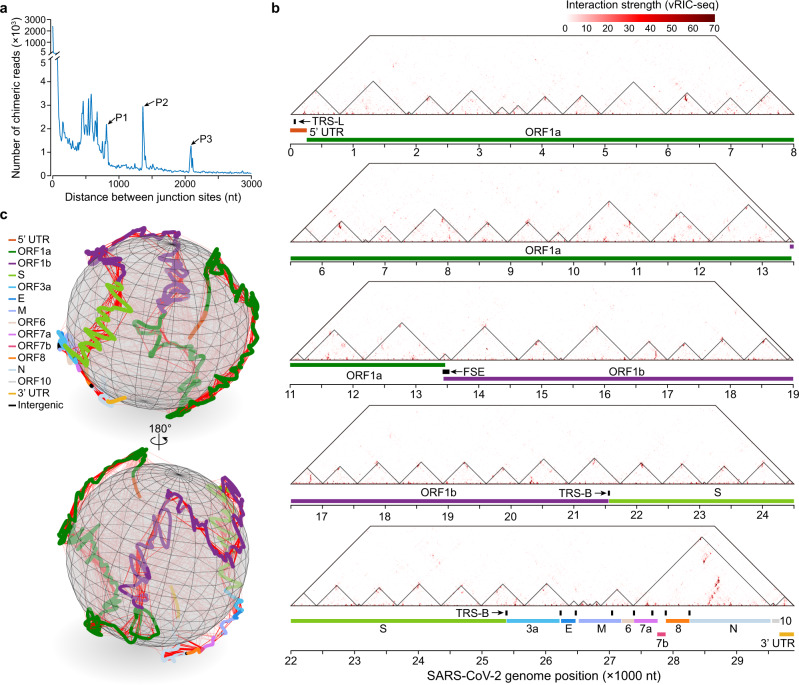
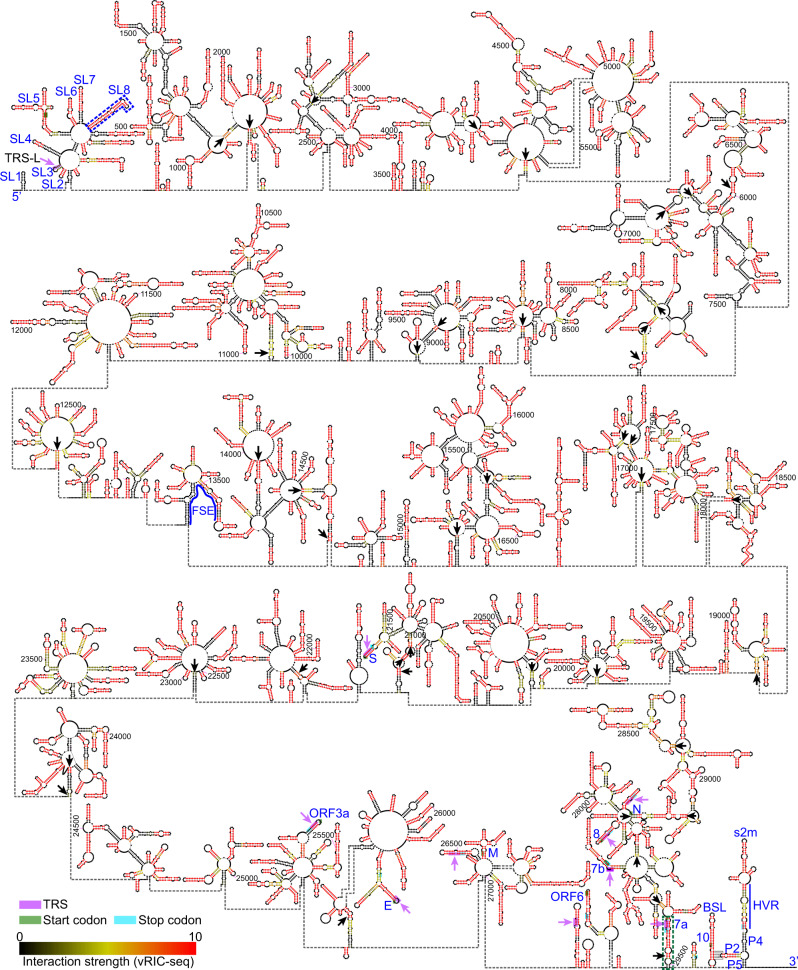
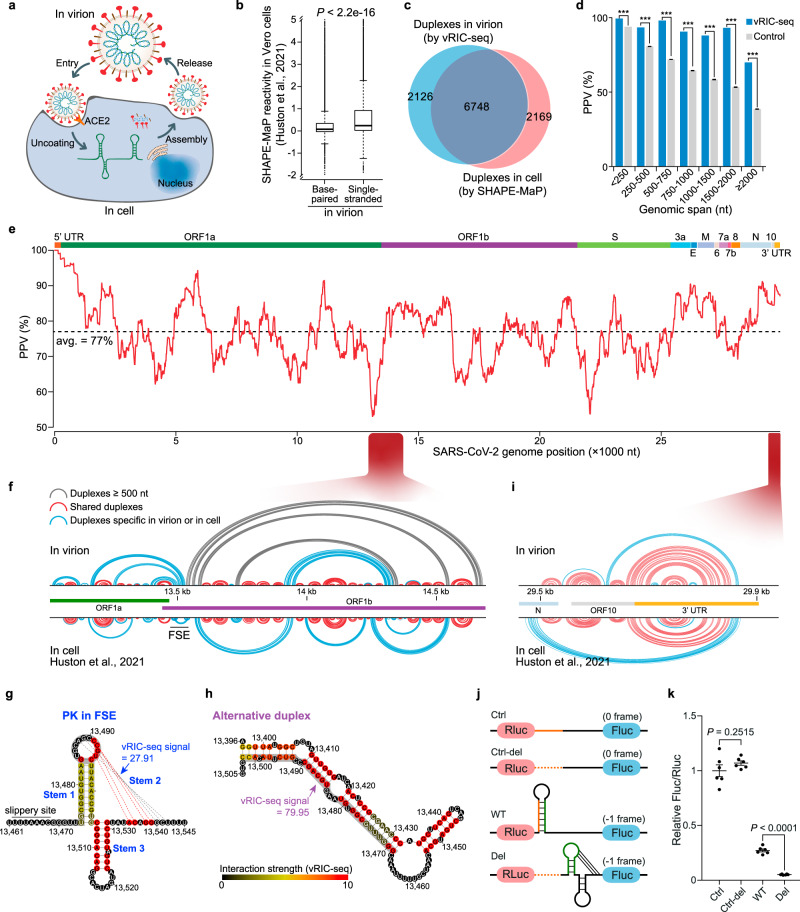
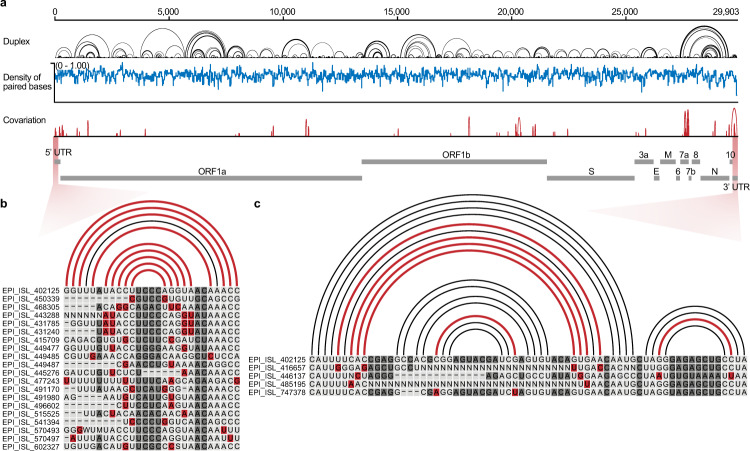
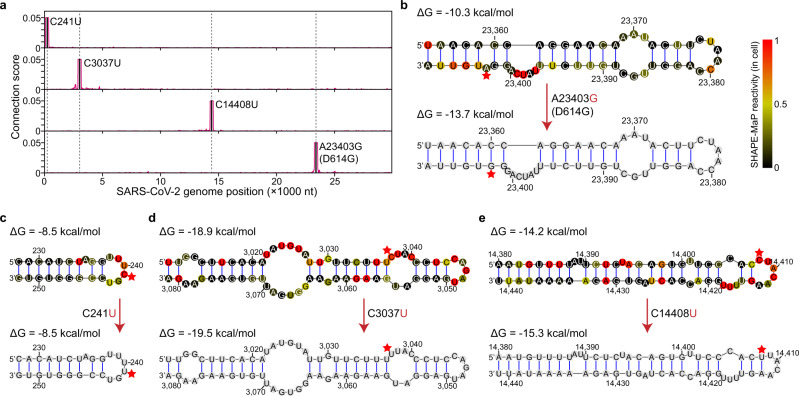
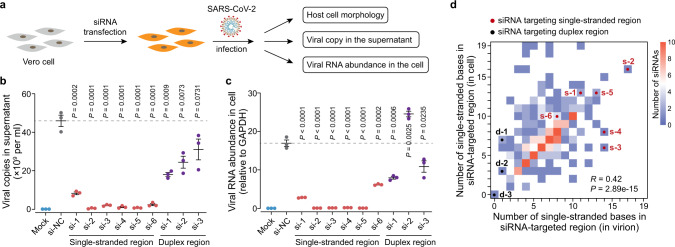
SARS-CoV-2 carries the largest single-stranded RNA genome and is the causal pathogen of the ongoing COVID-19 pandemic. How the SARS-CoV-2 RNA genome is folded in the virion remains unknown. To fill the knowledge gap and facilitate structure-based drug development, we develop a virion RNA in situ conformation sequencing technology, named vRIC-seq, for probing viral RNA genome structure unbiasedly. Using vRIC-seq data, we reconstruct the tertiary structure of the SARS-CoV-2 genome and reveal a surprisingly "unentangled globule" conformation. We uncover many long-range duplexes and higher-order junctions, both of which are under purifying selections and contribute to the sequential package of the SARS-CoV-2 genome. Unexpectedly, the D614G and the other two accompanying mutations may remodel duplexes into more stable forms. Lastly, the structure-guided design of potent small interfering RNAs can obliterate the SARS-CoV-2 in Vero cells. Overall, our work provides a framework for studying the genome structure, function, and dynamics of emerging deadly RNA viruses.
Conflict of interest statement
The authors declare no competing interests.
Figures
Similar articles
-
Cryo-EM and antisense targeting of the 28-kDa frameshift stimulation element from the SARS-CoV-2 RNA genome.Nat Struct Mol Biol. 2021 Sep;28(9):747-754. doi: 10.1038/s41594-021-00653-y. Epub 2021 Aug 23. Nat Struct Mol Biol. 2021. PMID: 34426697 Free PMC article.
-
Structure and Dynamics of RNA Guanine Quadruplexes in SARS-CoV-2 Genome. Original Strategies against Emerging Viruses.J Phys Chem Lett. 2021 Oct 28;12(42):10277-10283. doi: 10.1021/acs.jpclett.1c03071. Epub 2021 Oct 15. J Phys Chem Lett. 2021. PMID: 34652910
-
In vivo structure and dynamics of the SARS-CoV-2 RNA genome.Nat Commun. 2021 Sep 28;12(1):5695. doi: 10.1038/s41467-021-25999-1. Nat Commun. 2021. PMID: 34584097 Free PMC article.
-
Structure and regulation of coronavirus genomes: state-of-the-art and novel insights from SARS-CoV-2 studies.Biochem Soc Trans. 2021 Feb 26;49(1):341-352. doi: 10.1042/BST20200670. Biochem Soc Trans. 2021. PMID: 33367597 Free PMC article. Review.
-
SARS-CoV-2 Subgenomic RNAs: Characterization, Utility, and Perspectives.Viruses. 2021 Sep 24;13(10):1923. doi: 10.3390/v13101923. Viruses. 2021. PMID: 34696353 Free PMC article. Review.
Cited by
-
Epitopes recognition of SARS-CoV-2 nucleocapsid RNA binding domain by human monoclonal antibodies.iScience. 2024 Jan 19;27(2):108976. doi: 10.1016/j.isci.2024.108976. eCollection 2024 Feb 16. iScience. 2024. PMID: 38327783 Free PMC article.
-
Double-stranded RNA drives SARS-CoV-2 nucleocapsid protein to undergo phase separation at specific temperatures.Nucleic Acids Res. 2022 Aug 12;50(14):8168-8192. doi: 10.1093/nar/gkac596. Nucleic Acids Res. 2022. PMID: 35871289 Free PMC article.
-
Unveiling the nature's fruit basket to computationally identify Citrus sinensis csi-mir169-3p as a probable plant miRNA against Reference and Omicron SARS-CoV-2 genome.Comput Biol Med. 2022 Jul;146:105502. doi: 10.1016/j.compbiomed.2022.105502. Epub 2022 Apr 8. Comput Biol Med. 2022. PMID: 35605482 Free PMC article.
-
Inhibition of SARS-CoV-2 coronavirus proliferation by designer antisense-circRNAs.Nucleic Acids Res. 2021 Dec 2;49(21):12502-12516. doi: 10.1093/nar/gkab1096. Nucleic Acids Res. 2021. PMID: 34850109 Free PMC article.
-
Exploiting functional regions in the viral RNA genome as druggable entities.Elife. 2025 Jul 2;13:RP103923. doi: 10.7554/eLife.103923. Elife. 2025. PMID: 40600408 Free PMC article.
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
