

Metagenomic compendium of 189,680 DNA viruses from the human gut microbiome
- PMID: 34168315
- PMCID: PMC8241571
- DOI: 10.1038/s41564-021-00928-6
Metagenomic compendium of 189,680 DNA viruses from the human gut microbiome
Abstract
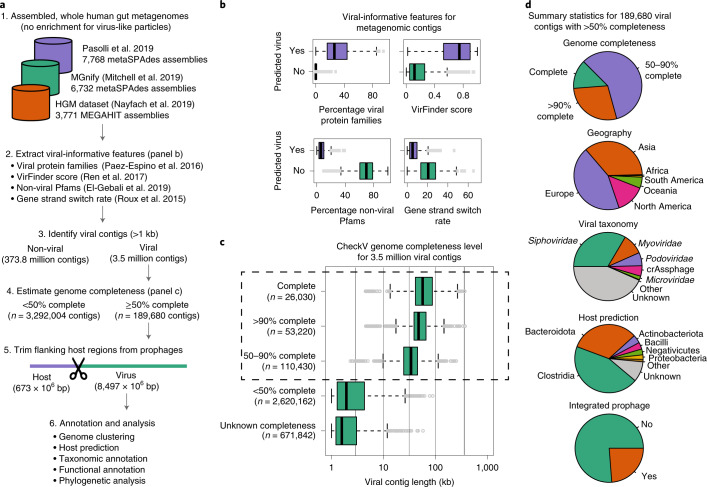
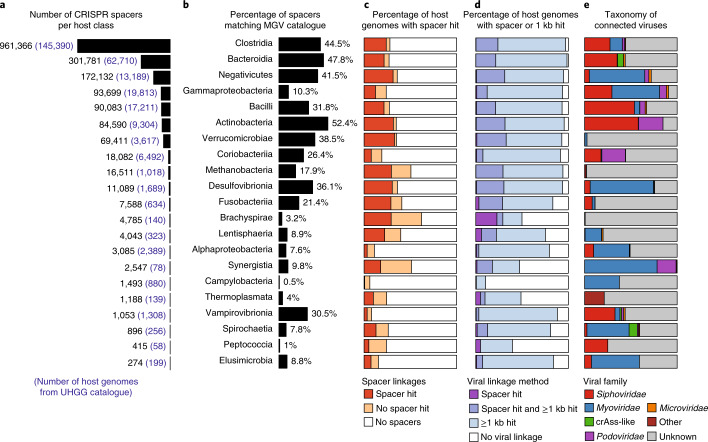
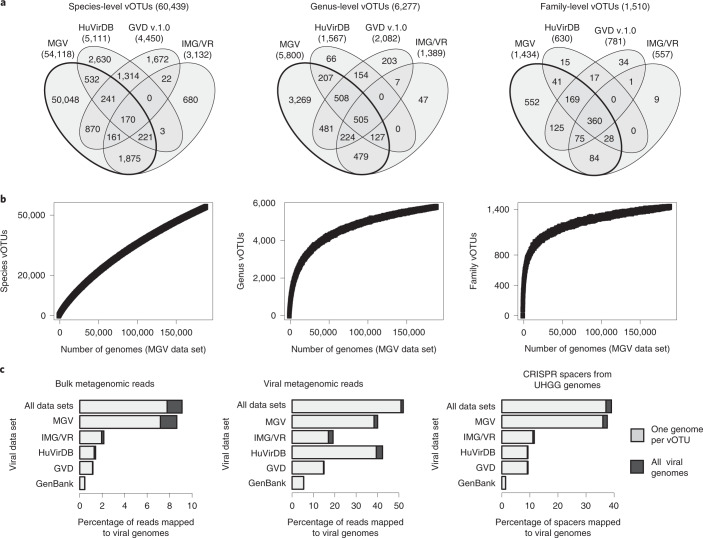
Bacteriophages have important roles in the ecology of the human gut microbiome but are under-represented in reference databases. To address this problem, we assembled the Metagenomic Gut Virus catalogue that comprises 189,680 viral genomes from 11,810 publicly available human stool metagenomes. Over 75% of genomes represent double-stranded DNA phages that infect members of the Bacteroidia and Clostridia classes. Based on sequence clustering we identified 54,118 candidate viral species, 92% of which were not found in existing databases. The Metagenomic Gut Virus catalogue improves detection of viruses in stool metagenomes and accounts for nearly 40% of CRISPR spacers found in human gut Bacteria and Archaea. We also produced a catalogue of 459,375 viral protein clusters to explore the functional potential of the gut virome. This revealed tens of thousands of diversity-generating retroelements, which use error-prone reverse transcription to mutate target genes and may be involved in the molecular arms race between phages and their bacterial hosts.
Conflict of interest statement
P.H. is a co-founder of Microba Life Sciences, which is a microbial genomics company developing microbiome-based diagnostics and therapeutics and offers metagenomic gut microbiome reports. All other authors declare no competing interests.
Figures







References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
