

Review
doi: 10.1093/hmg/ddab170.
Huntington's disease: nearly four decades of human molecular genetics
Affiliations
- PMID: 34169318
- PMCID: PMC8490011
- DOI: 10.1093/hmg/ddab170
Item in Clipboard
Review
Huntington's disease: nearly four decades of human molecular genetics
Hum Mol Genet.
.
Abstract
Huntington's disease (HD) is a devastating neurogenetic disorder whose familial nature and progressive course were first described in the 19th century but for which no disease-modifying treatment is yet available. Through the active participation of HD families, this disorder has acted as a flagship for the application of human molecular genetic strategies to identify disease genes, understand pathogenesis and identify rational targets for development of therapies.
© The Author(s) 2021. Published by Oxford University Press. All rights reserved. For Permissions, please email: journals.permissions@oup.com.
Figures
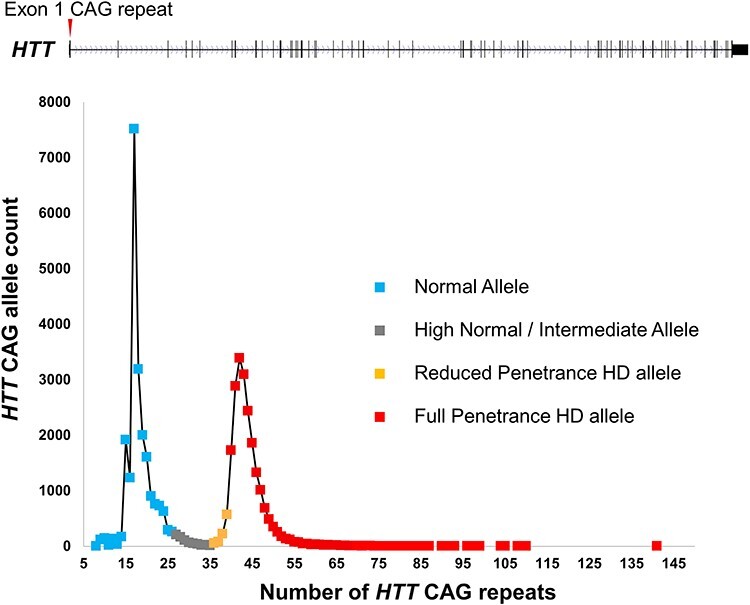
HTT CAG repeat distribution and relationship to HD. The 67-exon HTT gene and the location of its polymorphic CAG repeat (red arrow) in exon 1 are shown above a plot of the distribution of CAG allele lengths in >20 000 HD study participants (almost all HD heterozygotes) genotyped by the Molecular Neurogenetics Unit at the Massachusetts General Hospital. The distribution forms a continuum of allele lengths, with soft borders that distinguish allele lengths associated with phenotype. Normal alleles (approximately <27 CAGs), shown in blue, are not associated with a phenotype and are inherited in a stable manner. High normal alleles (shown in gray), sometimes called intermediate alleles (approximately >26 but < 36 CAGs), do not typically develop signs of HD but may show some degree of germline instability of the CAG repeat. Reduced penetrance alleles (roughly 36–39 CAGs and shown in orange) are associated with HD, but not all individuals with these alleles will develop clinical signs of the disorder. Full penetrance alleles (>39 CAGs, shown in red) are almost always associated with development of clinical signs and symptoms of HD. Of the expanded alleles (>35 CAGs) depicted, 4.4% are in the reduced penetrance range and 2% have >55 CAGs, in the range usually associated with very early onset of HD.
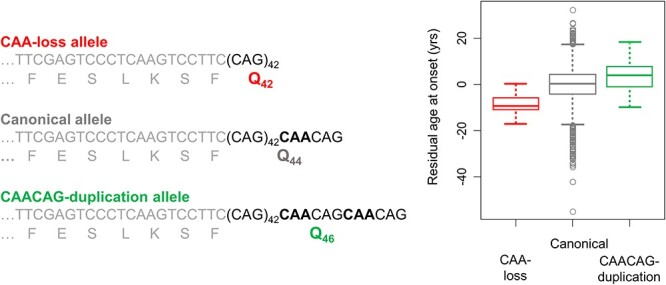
The timing of HD onset is driven by a property of CAG repeat length, not polyglutamine. Canonical HTT CAG expansion alleles associated with HD have repeats with a penultimate CAA interruption. Since both CAG and CAA are codons that specify glutamine, this results in a huntingtin protein with two more glutamines in its polyglutamine repeat than there are consecutive CAGs in the DNA. This figure depicts the effect of two minor variant alleles, one in which the CAA is lost (red) and one in which a second CAA results in effective duplication of the terminal CAACAG segment (green). To the left, CAA-loss, canonical and CAACAG-duplication alleles, all with pure CAG tracts of 43 units, are compared, showing that each specifies a different number of glutamines. On the right, a box plot of residual age at onset (i.e. the difference between observed age at onset and that expected based upon the uninterrupted CAG repeat length) for the set of participants in the GeM-HD GWAS studies shows that, despite having the longest polyglutamine length, those with CAACAG-duplication (N = 69) alleles do not show earlier onset. In contrast, the CAA-loss allele carriers (N = 21) show earlier onset than canonical (N = 7724; eight most frequent haplotypes) and CAACAG-duplication carriers despite having the shortest polyglutamine tracts.
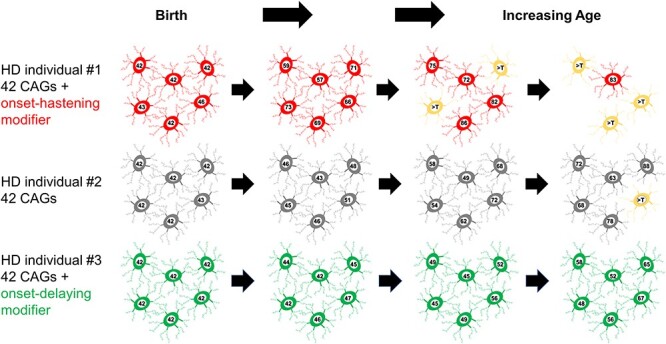
Genetic modification of the rate driver of HD pathogenesis. This figure provides an illustration of the phases of the two-component model of HD pathogenesis that has emerged from human molecular genetic studies. The timing of HD onset is driven by the uninterrupted CAG repeat length and is influenced by genetic modifiers that are associated with somatic expansion of trinucleotide repeats. Depicted are sets of medium-spiny striatal neurons at birth and with increasing age in individuals who inherited 42 uninterrupted HTT CAG repeats but with different modifier alleles. With progressive age, the CAG length shows variable degrees of expansion, with the greatest and least occurring in the individual with a strong onset-hastening modifier (red) and with a strong onset-delaying modifier (green), respectively. When the repeat in a given neuron reaches a threshold repeat length (noted as >T, a length that is currently not known but is assumed for this presentation to be >90 CAGs), toxic damage occurs through a mechanism that is not yet certain, resulting in neuronal dysfunction (yellow) and eventual death (disappearance).
References
-
- Huntington, G. (1872) On chorea. Med. Surg. Reporter of Philadelphia, 26, 317–321.
-
- Gusella, J.F., Wexler, N.S., Conneally, P.M., Naylor, S.L., Anderson, M.A., Tanzi, R.E., Watkins, P.C., Ottina, K., Wallace, M.R., Sakaguchi, A.Y. et al. (1983) A polymorphic DNA marker genetically linked to Huntington's disease. Nature, 306, 234–238. - PubMed
-
- Pericak-Vance, M.A., Conneally, P.M., Merritt, A.D., Roos, R., Norton, J.A., Jr. and Vance, J.M. (1978) Genetic linkage studies in Huntington disease. Cytogenet. Cell Genet., 22, 640–645. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
