

The AML microenvironment catalyzes a stepwise evolution to gilteritinib resistance
- PMID: 34171263
- PMCID: PMC8686208
- DOI: 10.1016/j.ccell.2021.06.003
The AML microenvironment catalyzes a stepwise evolution to gilteritinib resistance
Abstract
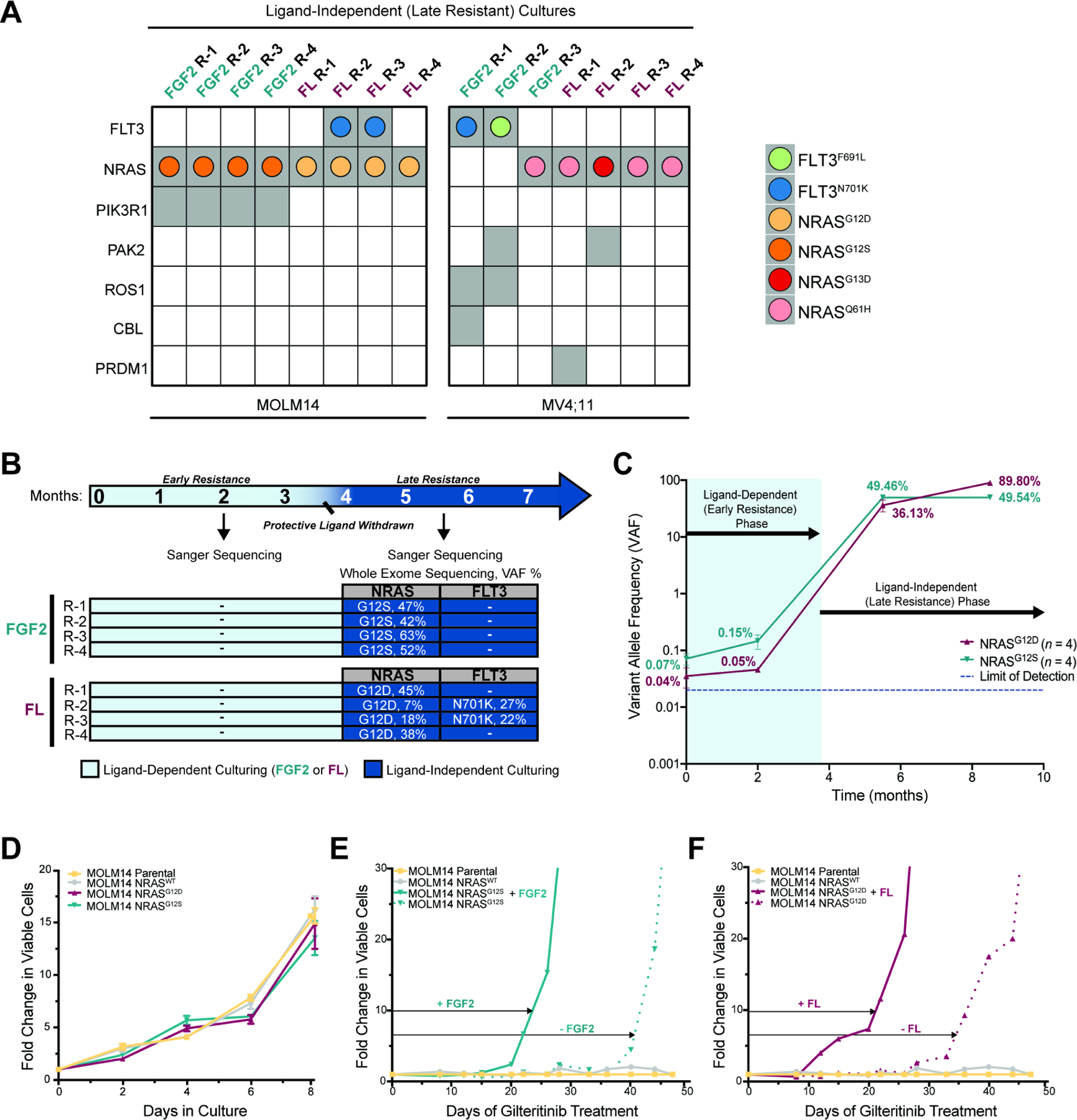
Our study details the stepwise evolution of gilteritinib resistance in FLT3-mutated acute myeloid leukemia (AML). Early resistance is mediated by the bone marrow microenvironment, which protects residual leukemia cells. Over time, leukemia cells evolve intrinsic mechanisms of resistance, or late resistance. We mechanistically define both early and late resistance by integrating whole-exome sequencing, CRISPR-Cas9, metabolomics, proteomics, and pharmacologic approaches. Early resistant cells undergo metabolic reprogramming, grow more slowly, and are dependent upon Aurora kinase B (AURKB). Late resistant cells are characterized by expansion of pre-existing NRAS mutant subclones and continued metabolic reprogramming. Our model closely mirrors the timing and mutations of AML patients treated with gilteritinib. Pharmacological inhibition of AURKB resensitizes both early resistant cell cultures and primary leukemia cells from gilteritinib-treated AML patients. These findings support a combinatorial strategy to target early resistant AML cells with AURKB inhibitors and gilteritinib before the expansion of pre-existing resistance mutations occurs.
Keywords: AML; Aurora kinase B; FLT3; NRAS; drug resistance; gilteritinib; lipid metabolism; quizartinib; tumor microenvironment; tyrosine kinase inhibitor.
Copyright © 2021 Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of interests B.J.D. potential competing interests—SAB: Aileron Therapeutics, Therapy Architects (ALLCRON), Cepheid, Vivid Biosciences, Celgene, RUNX1 Research Program, Novartis, Gilead Sciences (inactive), Monojul (inactive); SAB & Stock: Aptose Biosciences, Blueprint Medicines, EnLiven Therapeutics, Iterion Therapeutics, Third Coast Therapeutics, GRAIL (SAB inactive); Scientific Founder: MolecularMD (inactive, acquired by ICON); Board of Directors & Stock: Amgen, Vincera Pharma; Board of Directors: Burroughs Wellcome Fund, CureOne; Joint Steering Committee: Beat AML LLS; Founder: VB Therapeutics; Sponsored Research Agreement: EnLiven Therapeutics; Clinical Trial Funding: Novartis, Bristol-Myers Squibb, Pfizer; Royalties from Patent 6958335 (Novartis exclusive license) and OHSU and Dana-Farber Cancer Institute (one Merck exclusive license and one CytoImage, Inc., exclusive license). E.T. potential competing interests—Advisory Board/Consulting: Abbvie, Agios, Astellas, Daiichi-Sankyo; Clinical Trial Funding: Janssen, Incyte, LLS BeatAML. Stock options: Notable Labs. J.W.T. potential competing interests—research support: Agios, Aptose, Array, AstraZeneca, Constellation, Genentech, Gilead, Incyte, Janssen, Petra, Seattle Genetics, Syros, Tolero and Takeda. A.D. potential competing interests—founder: Omix Technologies, Inc., and Altis Biosciences, LLC; Consultant: Hemanext Inc. All other authors declare no potential competing interests.
Figures






Comment in
-
Stepwise evolution of therapy resistance in AML.Cancer Cell. 2021 Jul 12;39(7):904-906. doi: 10.1016/j.ccell.2021.06.004. Epub 2021 Jun 24. Cancer Cell. 2021. PMID: 34171262
References
-
- BATTULA VL, LE PM, SUN JC, NGUYEN K, YUAN B, ZHOU X, SONNYLAL S, MCQUEEN T, RUVOLO V, MICHEL KA, LING X, JACAMO R, SHPALL E, WANG Z, RAO A, AL-ATRASH G, KONOPLEVA M, DAVIS RE, HARRINGTON MA, CAHILL CW, BUESO-RAMOS C & ANDREEFF M 2017. AML-induced osteogenic differentiation in mesenchymal stromal cells supports leukemia growth. JCI Insight, 2. - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
