

An ancient viral epidemic involving host coronavirus interacting genes more than 20,000 years ago in East Asia
- PMID: 34171302
- PMCID: PMC8223470
- DOI: 10.1016/j.cub.2021.05.067
An ancient viral epidemic involving host coronavirus interacting genes more than 20,000 years ago in East Asia
Erratum in
-
An ancient viral epidemic involving host coronavirus interacting genes more than 20,000 years ago in East Asia.Curr Biol. 2021 Aug 23;31(16):3704. doi: 10.1016/j.cub.2021.07.052. Curr Biol. 2021. PMID: 34428405 Free PMC article. No abstract available.
Abstract
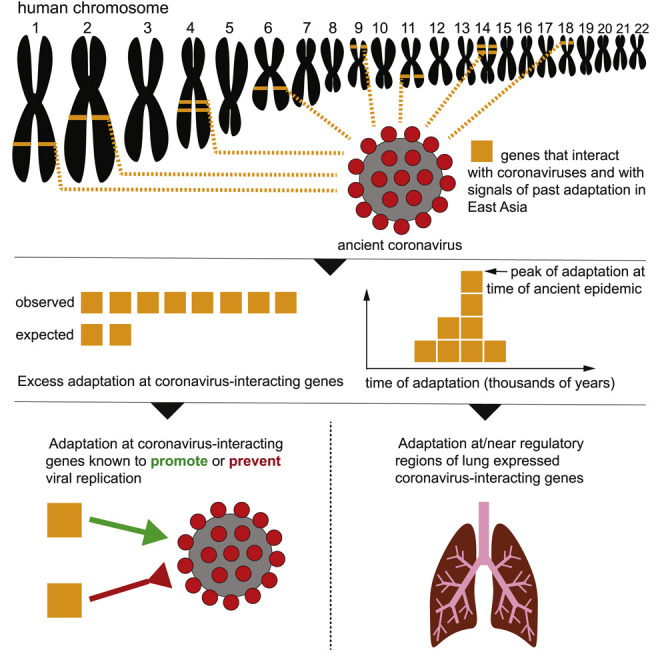
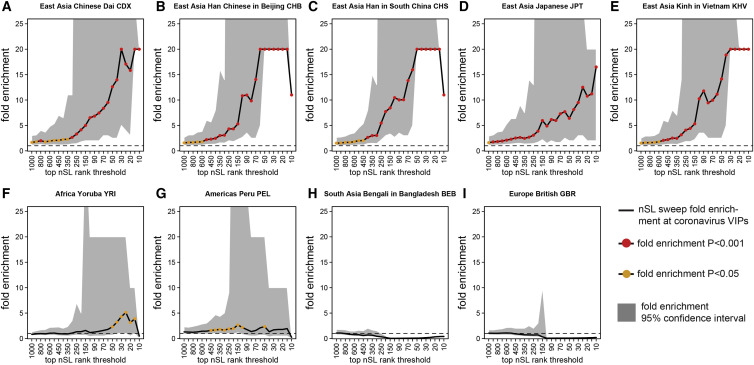
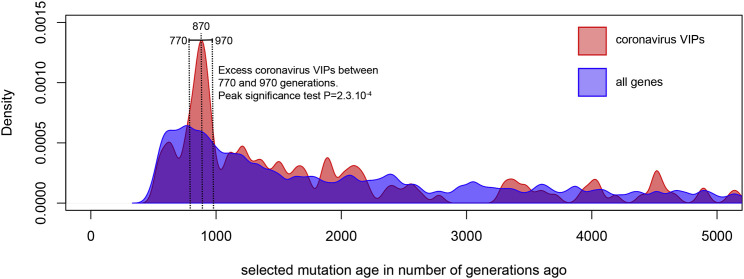
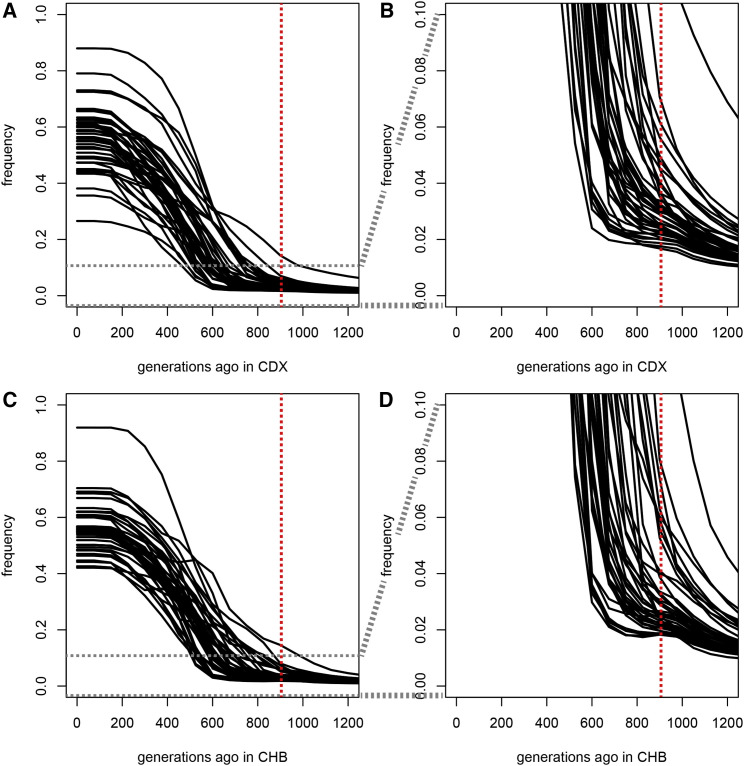
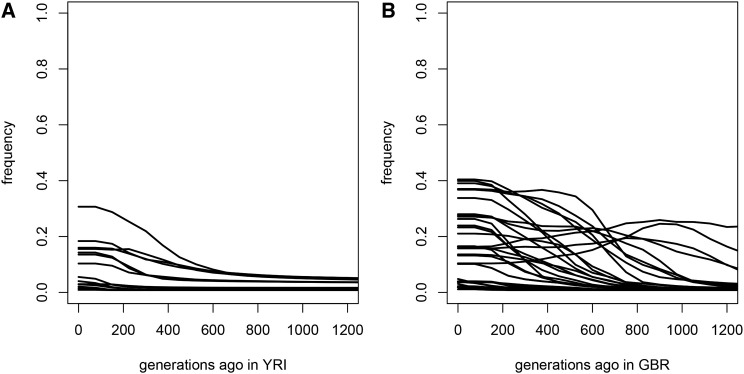
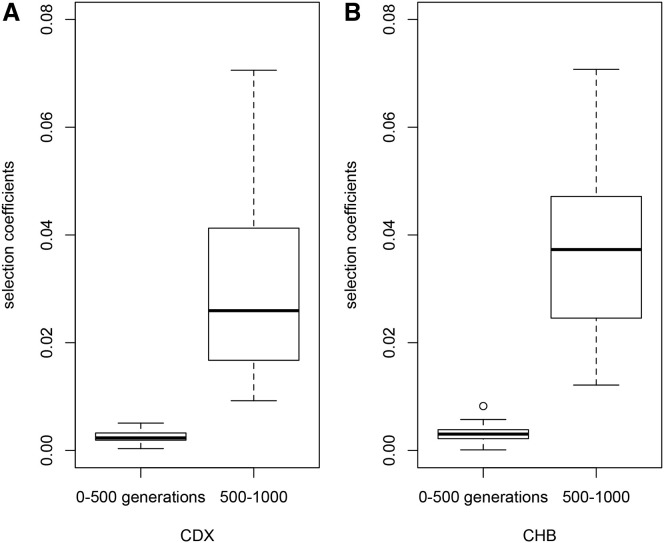
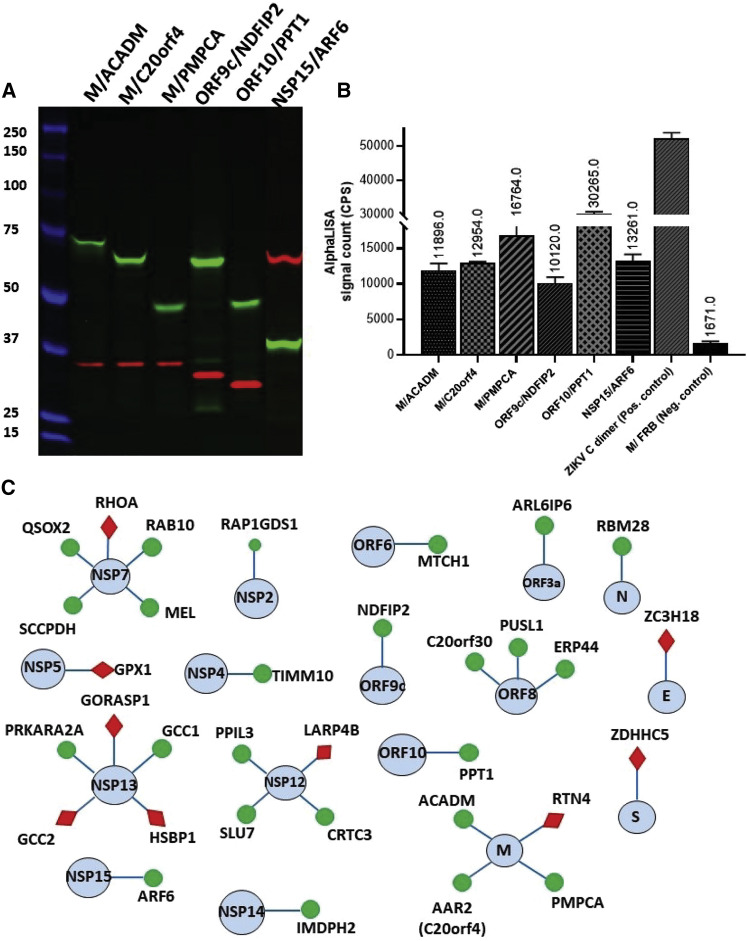
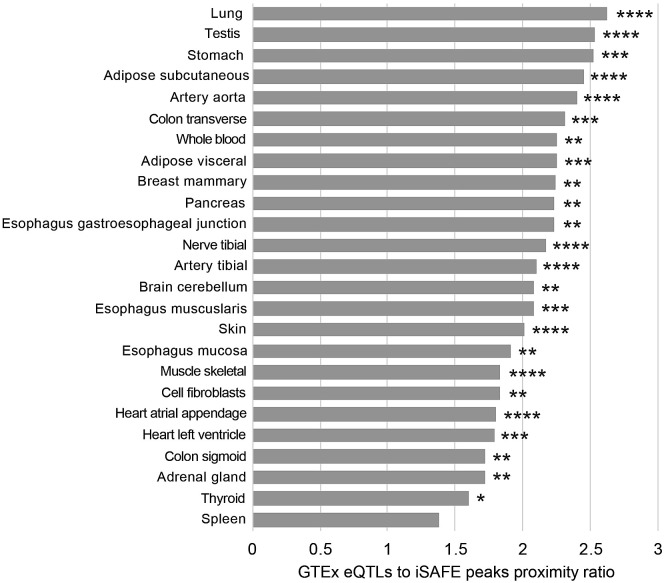
The current severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) pandemic has emphasized the vulnerability of human populations to novel viral pressures, despite the vast array of epidemiological and biomedical tools now available. Notably, modern human genomes contain evolutionary information tracing back tens of thousands of years, which may help identify the viruses that have impacted our ancestors-pointing to which viruses have future pandemic potential. Here, we apply evolutionary analyses to human genomic datasets to recover selection events involving tens of human genes that interact with coronaviruses, including SARS-CoV-2, that likely started more than 20,000 years ago. These adaptive events were limited to the population ancestral to East Asian populations. Multiple lines of functional evidence support an ancient viral selective pressure, and East Asia is the geographical origin of several modern coronavirus epidemics. An arms race with an ancient coronavirus, or with a different virus that happened to use similar interactions as coronaviruses with human hosts, may thus have taken place in ancestral East Asian populations. By learning more about our ancient viral foes, our study highlights the promise of evolutionary information to better predict the pandemics of the future. Importantly, adaptation to ancient viral epidemics in specific human populations does not necessarily imply any difference in genetic susceptibility between different human populations, and the current evidence points toward an overwhelming impact of socioeconomic factors in the case of coronavirus disease 2019 (COVID-19).
Keywords: ancient epidemics; coronaviruses; human genomes.
Copyright © 2021 The Authors. Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of interests The Krogan Laboratory has received research support from Vir Biotechnology and F. Hoffmann-La Roche. N.J.K. has consulting agreements with the Icahn School of Medicine at Mount Sinai, New York, Maze Therapeutics, and Interline Therapeutics; is a shareholder of Tenaya Therapeutics; and has received stocks from Maze Therapeutics and Interline Therapeutics. The other authors declare no competing interests.
Figures
Comment in
-
Coronavirus footprints.Nat Genet. 2021 Aug;53(8):1119. doi: 10.1038/s41588-021-00916-w. Nat Genet. 2021. PMID: 34363044 No abstract available.
References
-
- Hoffman C., Kamps B.S. Flying Publisher; 2003. SARS Reference.
-
- Balogun O.D., Bea V.J., Phillips E. Disparities in cancer outcomes due to COVID-19-a tale of 2 cities. JAMA Oncol. 2020;6:1531–1532. - PubMed
-
- Sattar N., McInnes I.B., McMurray J.J.V. Obesity is a risk factor for severe COVID-19 infection: multiple potential mechanisms. Circulation. 2020;142:4–6. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
