

Pharmacologic modulation of RNA splicing enhances anti-tumor immunity
- PMID: 34171309
- PMCID: PMC8684350
- DOI: 10.1016/j.cell.2021.05.038
Pharmacologic modulation of RNA splicing enhances anti-tumor immunity
Abstract
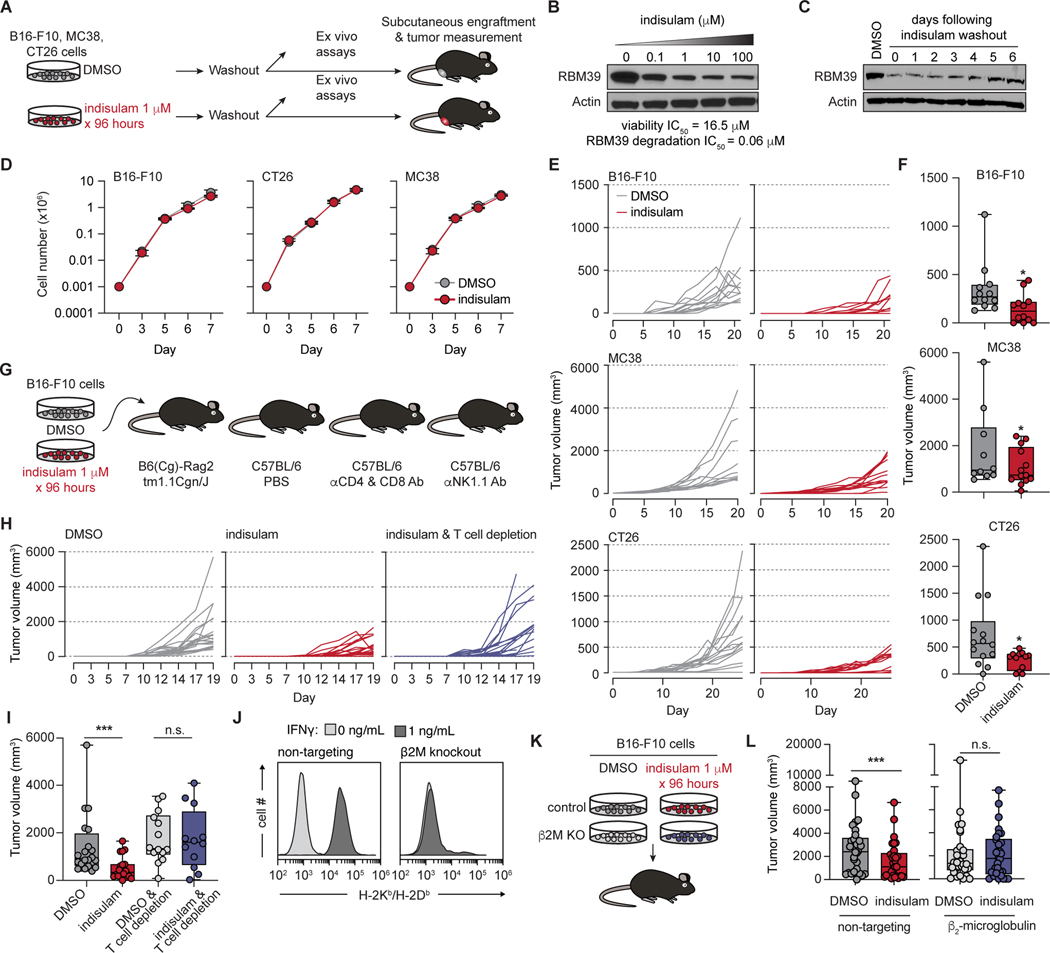
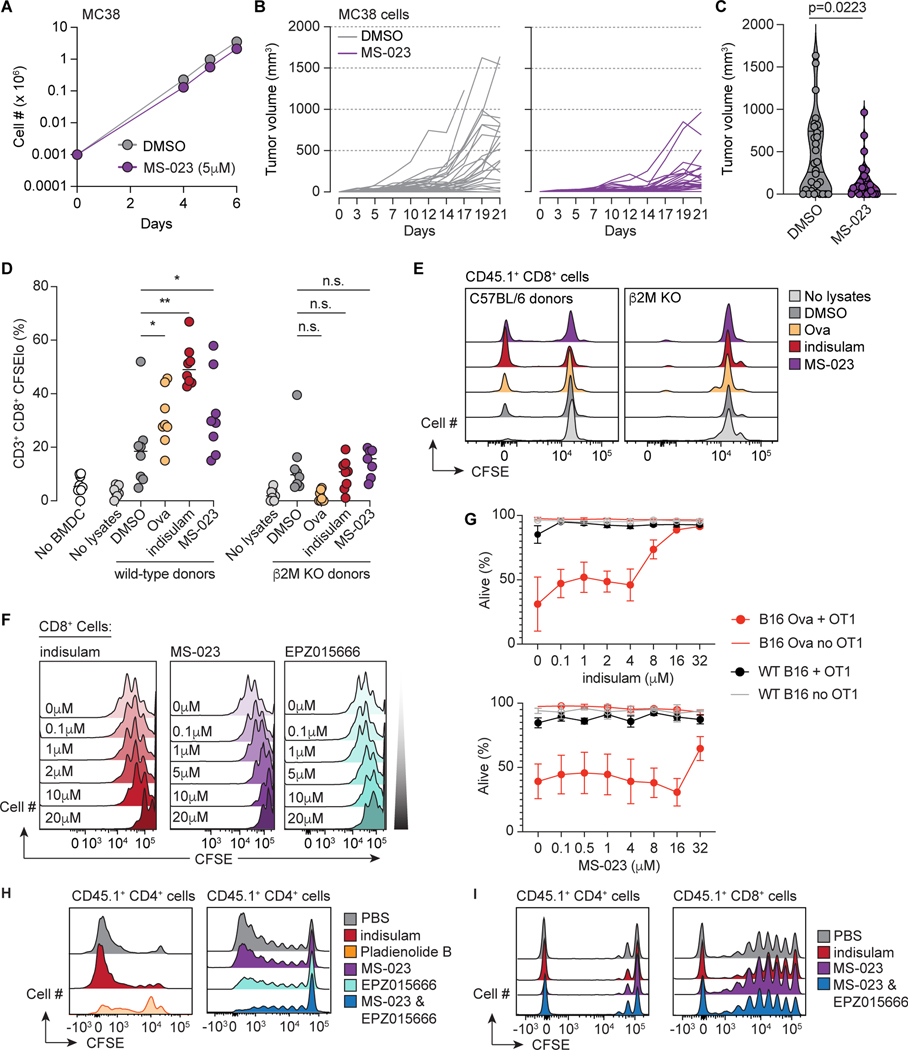
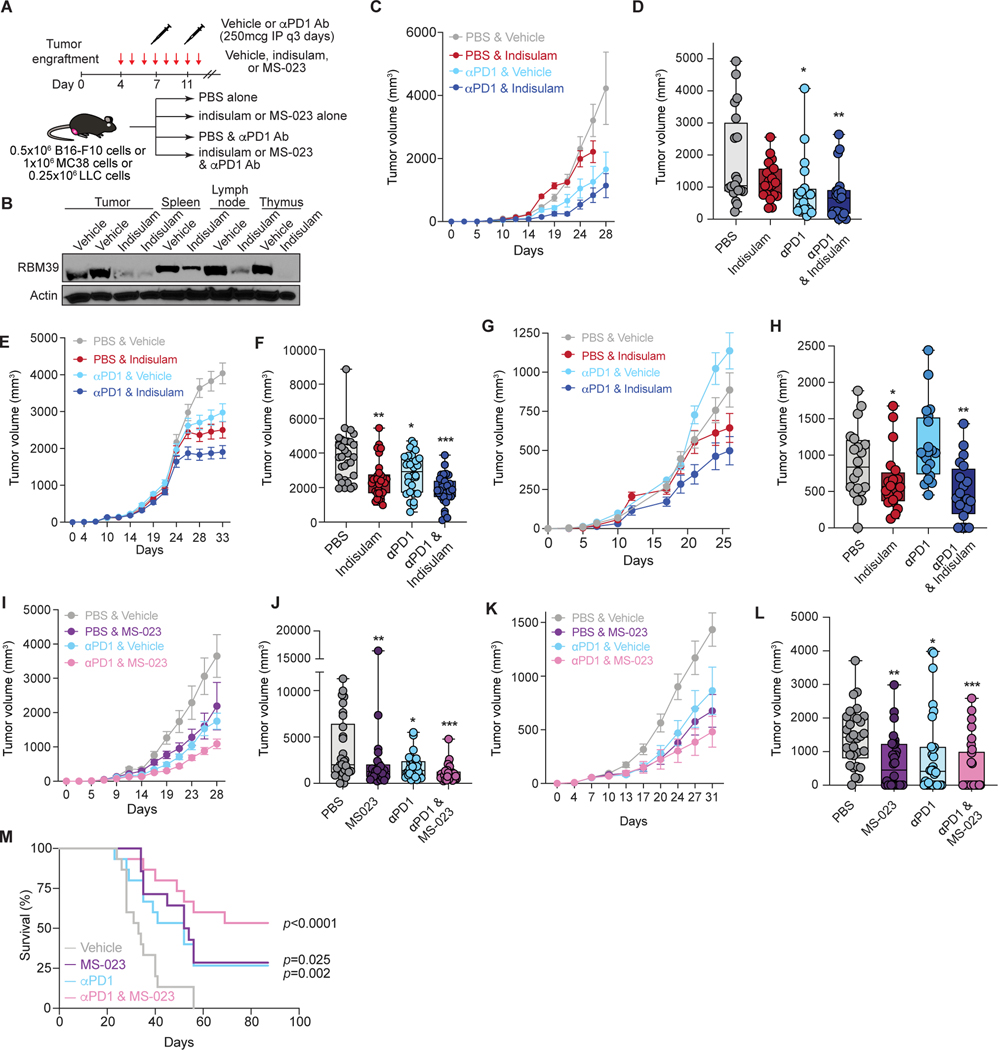
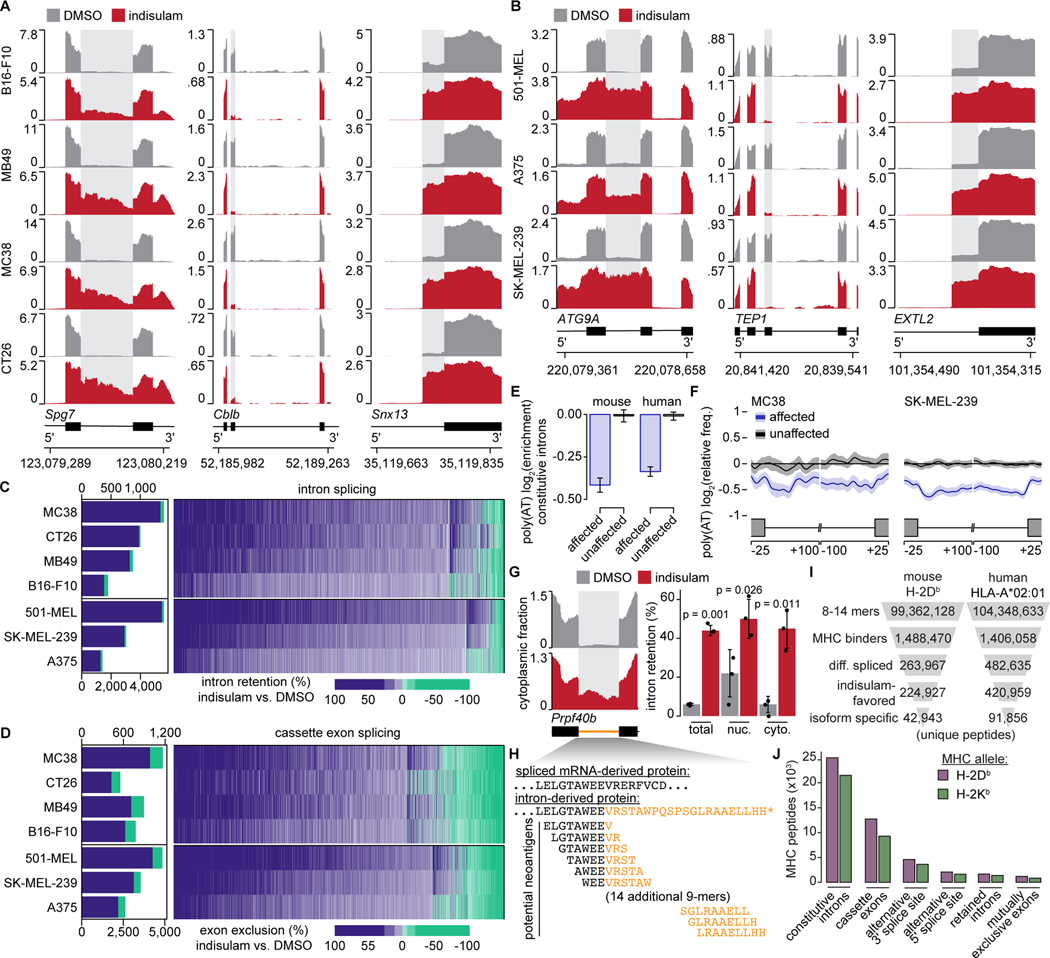
Although mutations in DNA are the best-studied source of neoantigens that determine response to immune checkpoint blockade, alterations in RNA splicing within cancer cells could similarly result in neoepitope production. However, the endogenous antigenicity and clinical potential of such splicing-derived epitopes have not been tested. Here, we demonstrate that pharmacologic modulation of splicing via specific drug classes generates bona fide neoantigens and elicits anti-tumor immunity, augmenting checkpoint immunotherapy. Splicing modulation inhibited tumor growth and enhanced checkpoint blockade in a manner dependent on host T cells and peptides presented on tumor MHC class I. Splicing modulation induced stereotyped splicing changes across tumor types, altering the MHC I-bound immunopeptidome to yield splicing-derived neoepitopes that trigger an anti-tumor T cell response in vivo. These data definitively identify splicing modulation as an untapped source of immunogenic peptides and provide a means to enhance response to checkpoint blockade that is readily translatable to the clinic.
Keywords: PD1; PRMTs; RBM39; RNA splicing; immune checkpoint blockade; immunopeptidome; immunotherapy; neoantigens; neoepitopes; splicing.
Copyright © 2021 Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of interests O.A.-W. has served as a consultant for H3B Biomedicine, Foundation Medicine, Merck, Prelude Therapeutics, and Janssen, and is on the scientific advisory board of Envisagenics, Pfizer Boulder, and AIChemy. O.A.-W. has received prior research funding from Loxo Oncology and H3 Biomedicine unrelated to this work. S.X.L. has served as a consultant (uncompensated) for PTC Therapeutics. B.R. has served as a consultant for Bayer and Roche. D.Z. reports clinical research support to his institution from Astra Zeneca, Plexxikon, and Genentech and personal/consultancy fees from Merck, Synlogic Therapeutics, Tesaro, Bristol Myers Squibb (BMS), Genentech, Xencor, Memgen, Calidi Biotherapeutics, and Agenus. L.A.D. is a member of the board of directors of Personal Genome Diagnostics (PGDx) and Jounce Therapeutics. L.A.D. is a compensated consultant to PGDx, 4Paws (PetDx), Innovatus CP, Se’er, Delfi, Kinnate, and Neophore. L.A.D. is an uncompensated consultant for, but has received clinical trial support from, Merck. L.A.D. holds equity in PGDx, Jounce, Se’er, Delfi, Kinnate, and Neophore and divested equity in Thrive Earlier Detection in 2021. His spouse holds equity in Amgen. J.D.W. is a consultant for Amgen, Apricity, Arsenal IO, Ascentage Pharma, AstraZeneca, Astellas, Boehringer Ingelheim, BMS, Chugai, Dragonfly, F Star, Eli Lilly, Georgiamune, IMVAQ, Merck, Polynoma, Psioxus, Recepta, Trieza, Truvax, Sellas, and Werewolf Therapeutics. J.D.W. has grant/research support from BMS and Sephora. J.D.W. reports equity in Tizona Pharmaceuticals, Imvaq, Beigene, Linneaus, Apricity, Arsenal IO, and Georgiamune. T.M. is an inventor on patents involving the use of anti-PD-1 antibodies. T.M. is a consultant for Immunos Therapeutics and Pfizer. T.M. is a cofounder of and equity holder in IMVAQ. T.M. receives research funding from BMS, Surface Oncology, Kyn Therapeutics, Infinity Pharmaceuticals, Peregrine Pharmaceuticals, Adaptive Biotechnologies, Leap Therapeutics, and Aprea Therapeutics. S.X.L., O.A.-W., and R.K.B. are inventors on a patent application submitted by FHCRC related to this work.
Figures
Comment in
-
Pharmacologic RNA splicing modulation: a novel mechanism to enhance neoantigen-directed anti-tumor immunity and immunotherapy response.Signal Transduct Target Ther. 2021 Oct 28;6(1):373. doi: 10.1038/s41392-021-00789-9. Signal Transduct Target Ther. 2021. PMID: 34711802 Free PMC article. No abstract available.
-
RNA Splicing and Immune-Checkpoint Inhibition.N Engl J Med. 2021 Nov 4;385(19):1807-1809. doi: 10.1056/NEJMcibr2110736. N Engl J Med. 2021. PMID: 34731541 No abstract available.
-
RNA splicing meets anti-tumor immunity.Nat Cancer. 2021 Dec;2(12):1287. doi: 10.1038/s43018-021-00309-2. Nat Cancer. 2021. PMID: 35121911 No abstract available.
References
-
- Chan-Penebre E, Kuplast KG, Majer CR, Boriack-Sjodin PA, Wigle TJ, Johnston LD, Rioux N, Munchhof MJ, Jin L, Jacques SL, et al. (2015). A selective inhibitor of PRMT5 with in vivo and in vitro potency in MCL models. Nat Chem Biol 11, 432–437. - PubMed
-
- De Bruijn ML, Schumacher TN, Nieland JD, Ploegh HL, Kast WM, and Melief CJ (1991). Peptide loading of empty major histocompatibility complex molecules on RMA-S cells allows the induction of primary cytotoxic T lymphocyte responses. Eur J Immunol 21, 2963–2970. - PubMed
-
- De Silva AD, Boesteanu A, Song R, Nagy N, Harhaj E, Harding CV, and Joyce S. (1999). Thermolabile H-2Kb molecules expressed by transporter associated with antigen processing-deficient RMA-S cells are occupied by low-affinity peptides. J Immunol 163, 4413–4420. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
