

Genetic variation in recombination rate in the pig
- PMID: 34171988
- PMCID: PMC8235837
- DOI: 10.1186/s12711-021-00643-0
Genetic variation in recombination rate in the pig
Abstract
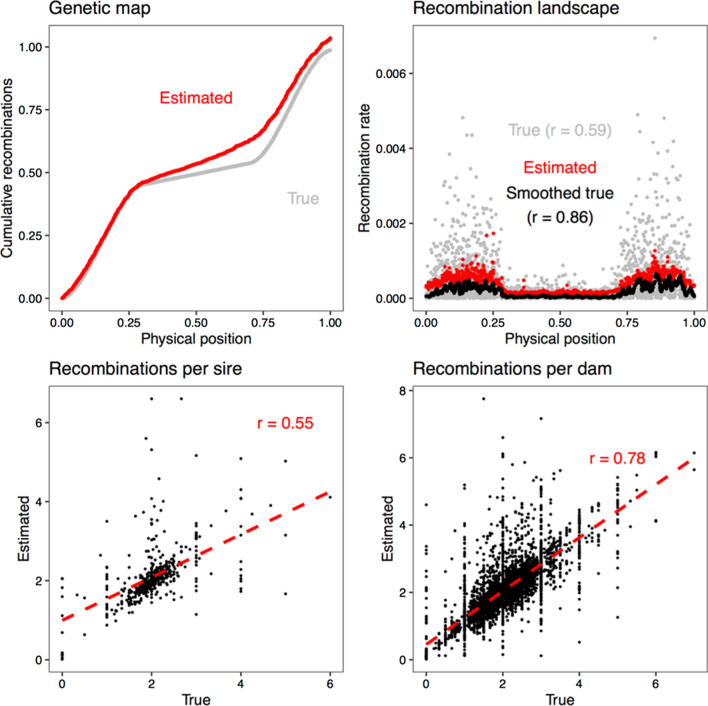
Background: Meiotic recombination results in the exchange of genetic material between homologous chromosomes. Recombination rate varies between different parts of the genome, between individuals, and is influenced by genetics. In this paper, we assessed the genetic variation in recombination rate along the genome and between individuals in the pig using multilocus iterative peeling on 150,000 individuals across nine genotyped pedigrees. We used these data to estimate the heritability of recombination and perform a genome-wide association study of recombination in the pig.
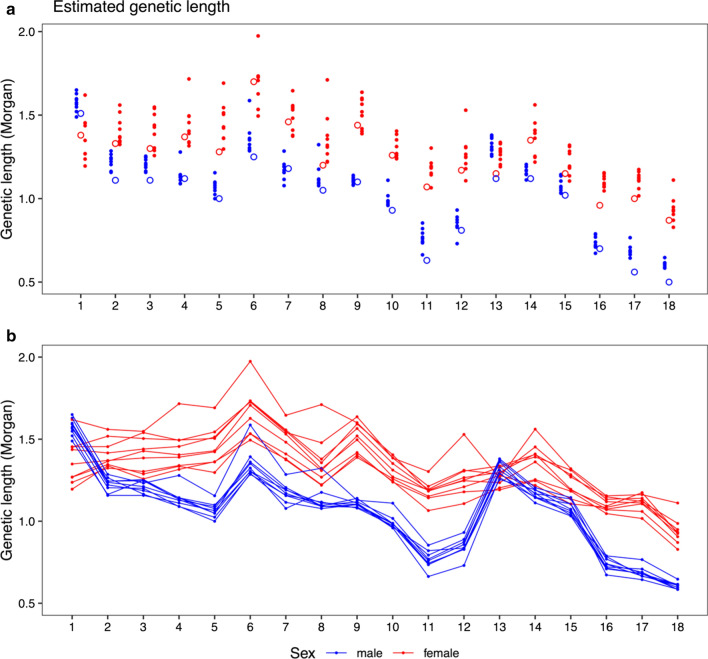
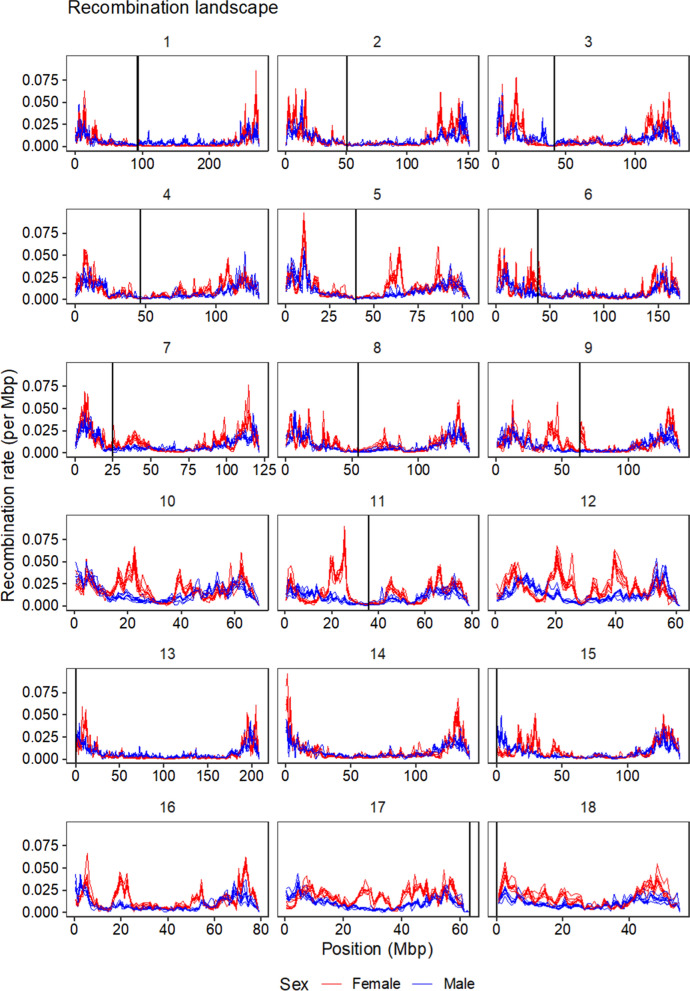
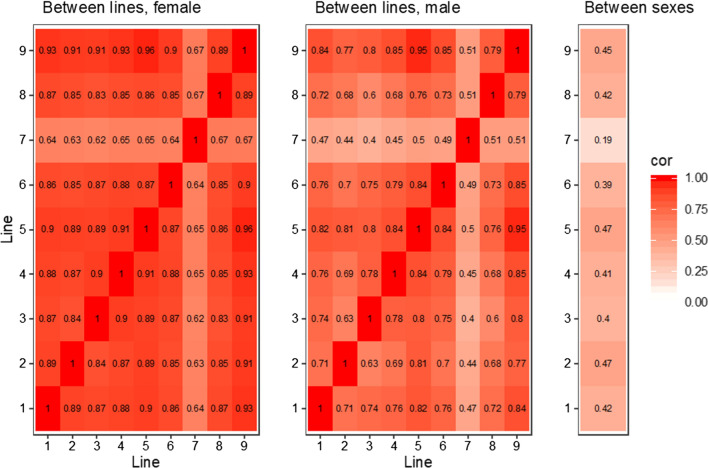
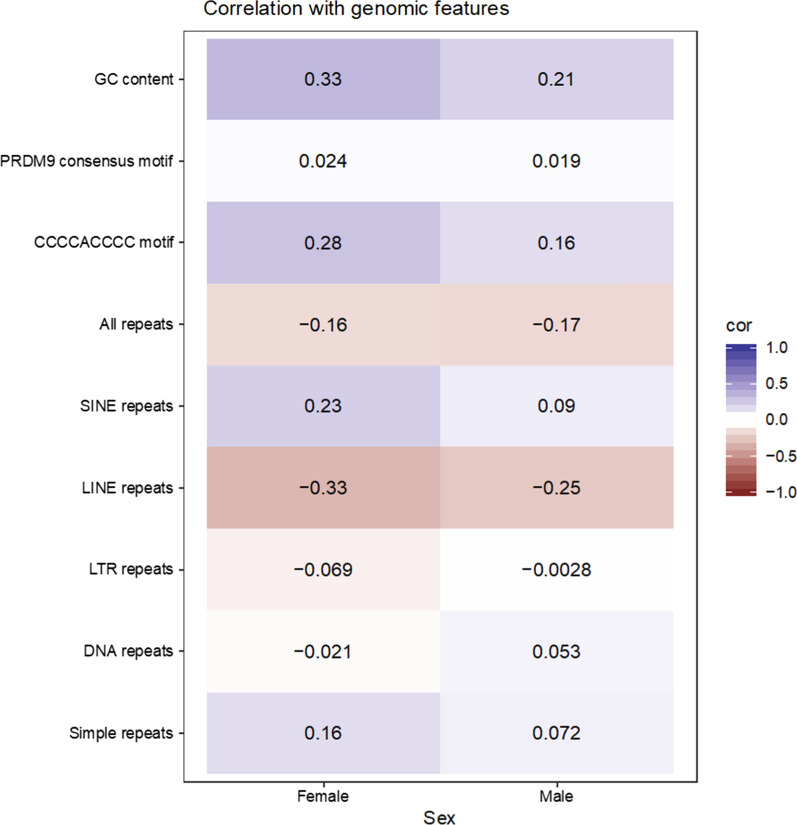
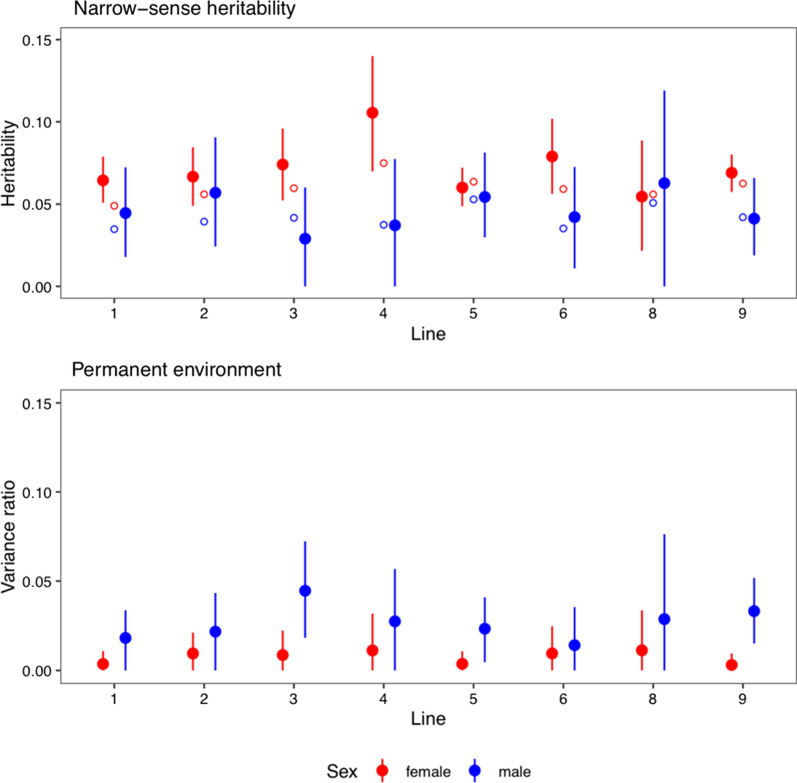
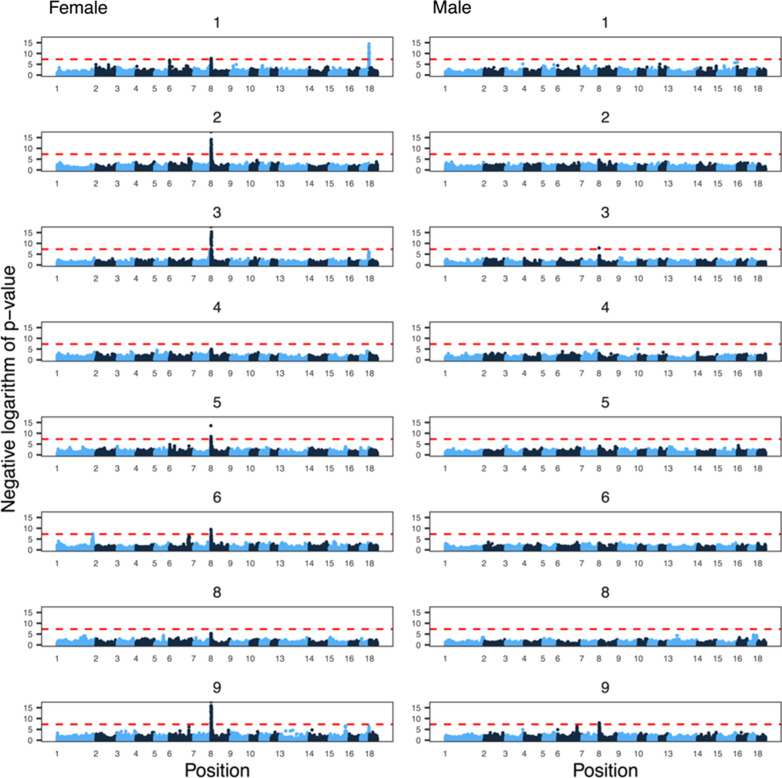
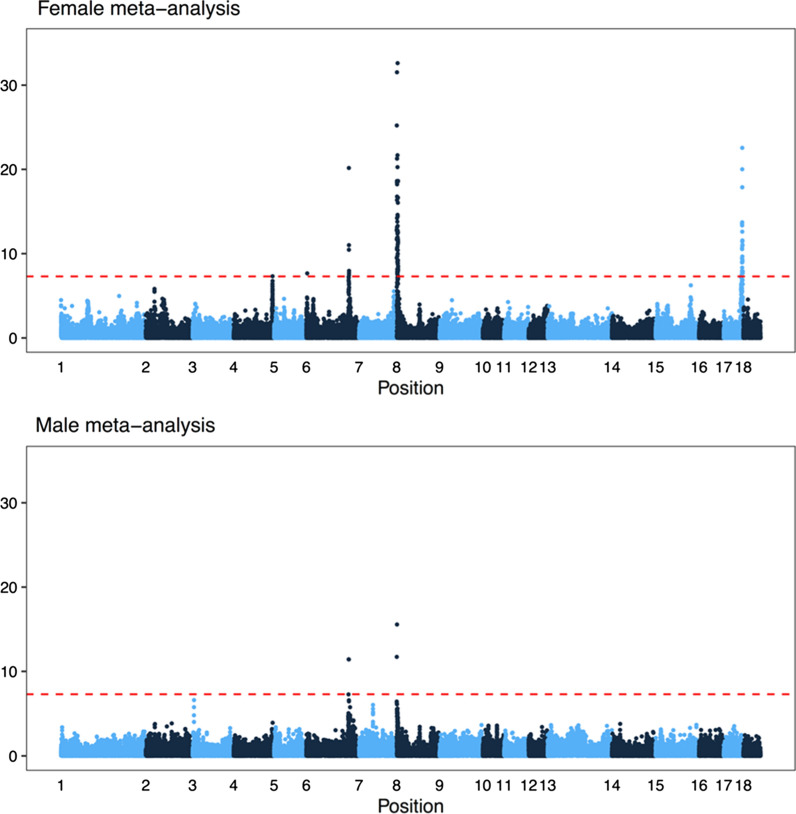
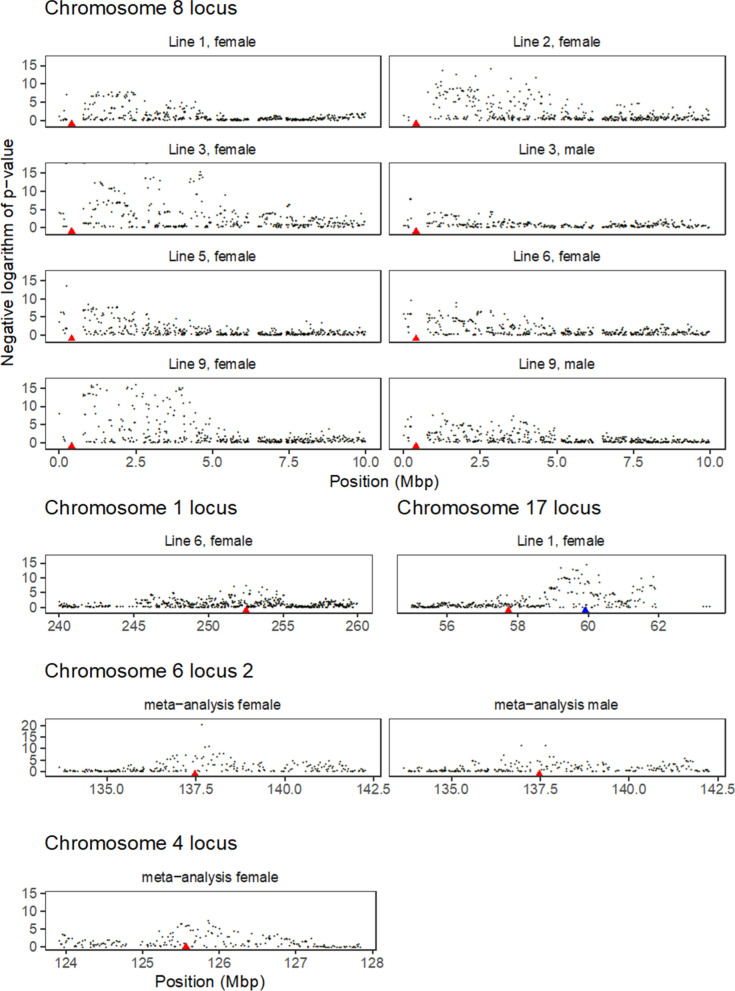
Results: Our results confirmed known features of the recombination landscape of the pig genome, including differences in genetic length of chromosomes and marked sex differences. The recombination landscape was repeatable between lines, but at the same time, there were differences in average autosome-wide recombination rate between lines. The heritability of autosome-wide recombination rate was low but not zero (on average 0.07 for females and 0.05 for males). We found six genomic regions that are associated with recombination rate, among which five harbour known candidate genes involved in recombination: RNF212, SHOC1, SYCP2, MSH4 and HFM1.
Conclusions: Our results on the variation in recombination rate in the pig genome agree with those reported for other vertebrates, with a low but nonzero heritability, and the identification of a major quantitative trait locus for recombination rate that is homologous to that detected in several other species. This work also highlights the utility of using large-scale livestock data to understand biological processes.
Conflict of interest statement
The authors declare that they have no competing interests.
Figures
References
MeSH terms
Grants and funding
- BBS/E/D/30002275/Biotechnology and Biological Sciences Research Council (GB)
- BB/N015339/1/Biotechnology and Biological Sciences Research Council (GB)
- BB/L020467/1/BB_/Biotechnology and Biological Sciences Research Council/United Kingdom
- BB/M009254/1/BB_/Biotechnology and Biological Sciences Research Council/United Kingdom
- 2016-01386/Svenska Forskningsrådet Formas
LinkOut - more resources
Full Text Sources
