

Loss-of-function variants in DNM1 cause a specific form of developmental and epileptic encephalopathy only in biallelic state
- PMID: 34172529
- PMCID: PMC9132866
- DOI: 10.1136/jmedgenet-2021-107769
Loss-of-function variants in DNM1 cause a specific form of developmental and epileptic encephalopathy only in biallelic state
Abstract
Background: Developmental and epileptic encephalopathies (DEEs) represent a group of severe neurological disorders characterised by an onset of refractory seizures during infancy or early childhood accompanied by psychomotor developmental delay or regression. DEEs are genetically heterogeneous with, to date, more than 80 different genetic subtypes including DEE31 caused by heterozygous missense variants in DNM1.
Methods: We performed a detailed clinical characterisation of two unrelated patients with DEE and used whole-exome sequencing to identify causative variants in these individuals. The identified variants were tested for cosegregation in the respective families.
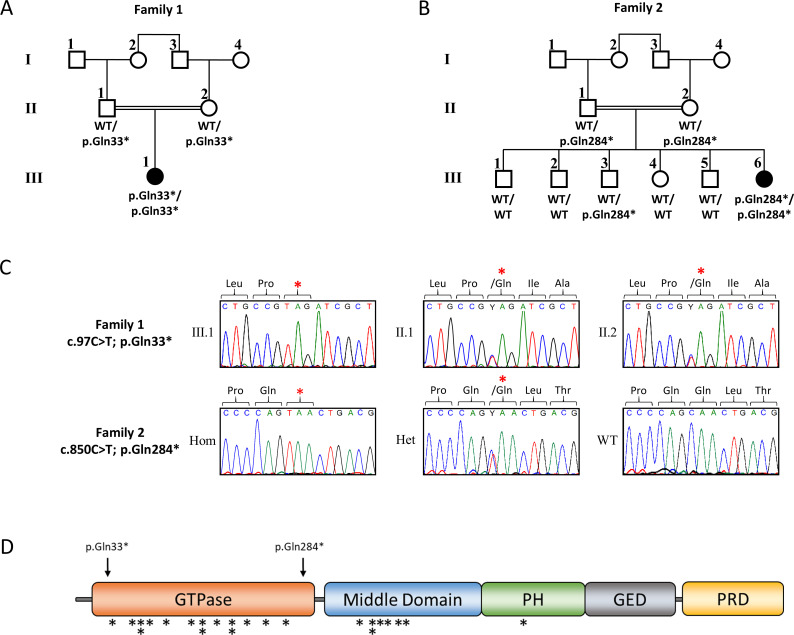
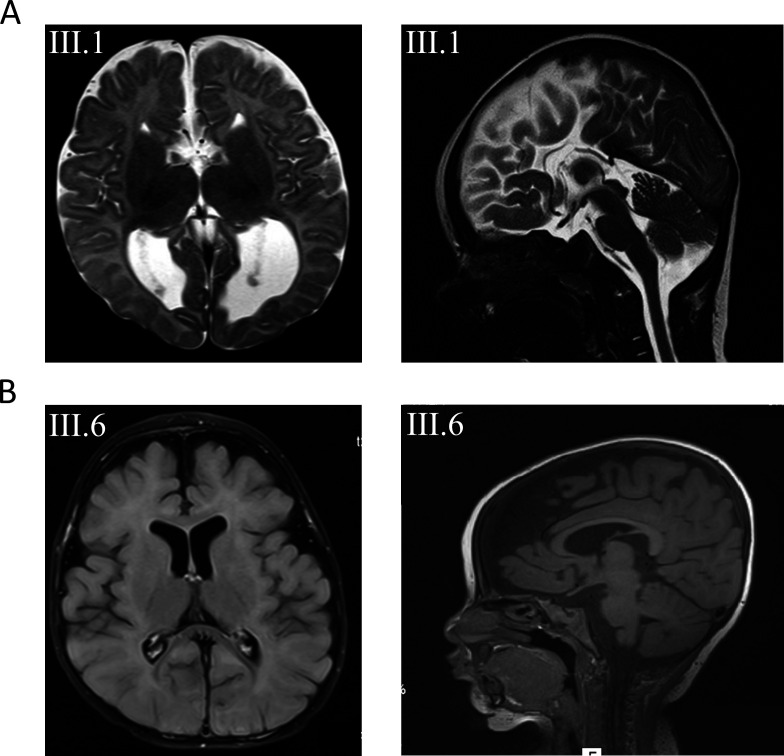
Results: We excluded pathogenic variants in known, DEE-associated genes. We identified homozygous nonsense variants, c.97C>T; p.(Gln33*) in family 1 and c.850C>T; p.(Gln284*) in family 2, in the DNM1 gene, indicating that biallelic, loss-of-function pathogenic variants in DNM1 cause DEE.
Conclusion: Our finding that homozygous, loss-of-function variants in DNM1 cause DEE expands the spectrum of pathogenic variants in DNM1. All parents who were heterozygous carriers of the identified loss-of-function variants were healthy and did not show any clinical symptoms, indicating that the type of mutation in DNM1 determines the pattern of inheritance.
Keywords: Pediatrics; epilepsy; genetics; nervous System Diseases.
© Author(s) (or their employer(s)) 2022. Re-use permitted under CC BY-NC. No commercial re-use. See rights and permissions. Published by BMJ.
Conflict of interest statement
Competing interests: None declared.
Figures
References
-
- Steward CA, Roovers J, Suner M-M, Gonzalez JM, Uszczynska-Ratajczak B, Pervouchine D, Fitzgerald S, Viola M, Stamberger H, Hamdan FF, Ceulemans B, Leroy P, Nava C, Lepine A, Tapanari E, Keiller D, Abbs S, Sanchis-Juan A, Grozeva D, Rogers AS, Diekhans M, Guigó R, Petryszak R, Minassian BA, Cavalleri G, Vitsios D, Petrovski S, Harrow J, Flicek P, Lucy Raymond F, Lench NJ, Jonghe PD, Mudge JM, Weckhuysen S, Sisodiya SM, Frankish A. Re-annotation of 191 developmental and epileptic encephalopathy-associated genes unmasks de novo variants in SCN1A. NPJ Genom Med 2019;4:1–11. 10.1038/s41525-019-0106-7 - DOI - PMC - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases