

Predicting genotoxicity of viral vectors for stem cell gene therapy using gene expression-based machine learning
- PMID: 34174440
- PMCID: PMC8636173
- DOI: 10.1016/j.ymthe.2021.06.017
Predicting genotoxicity of viral vectors for stem cell gene therapy using gene expression-based machine learning
Abstract
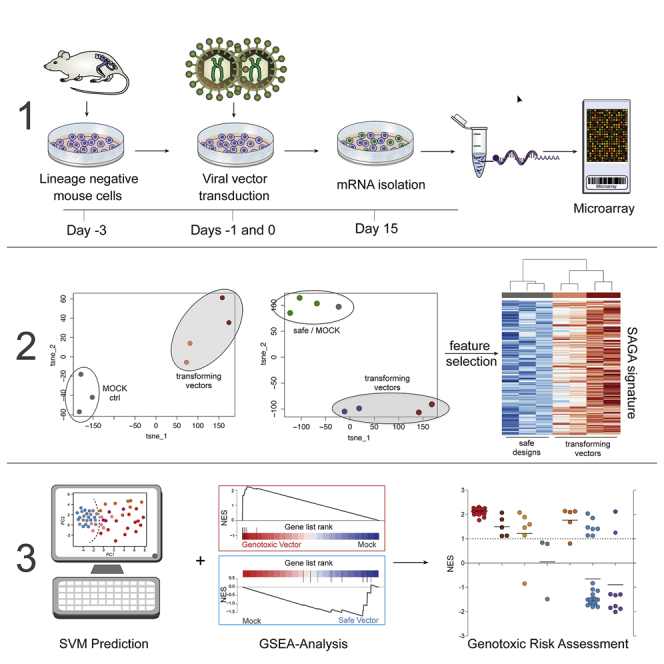
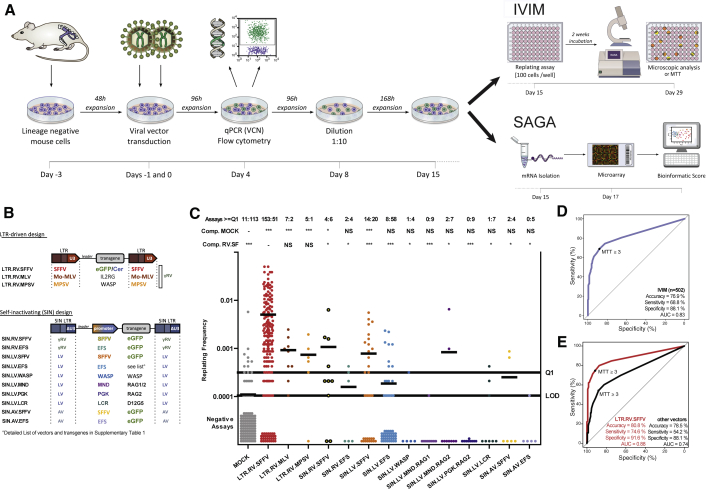
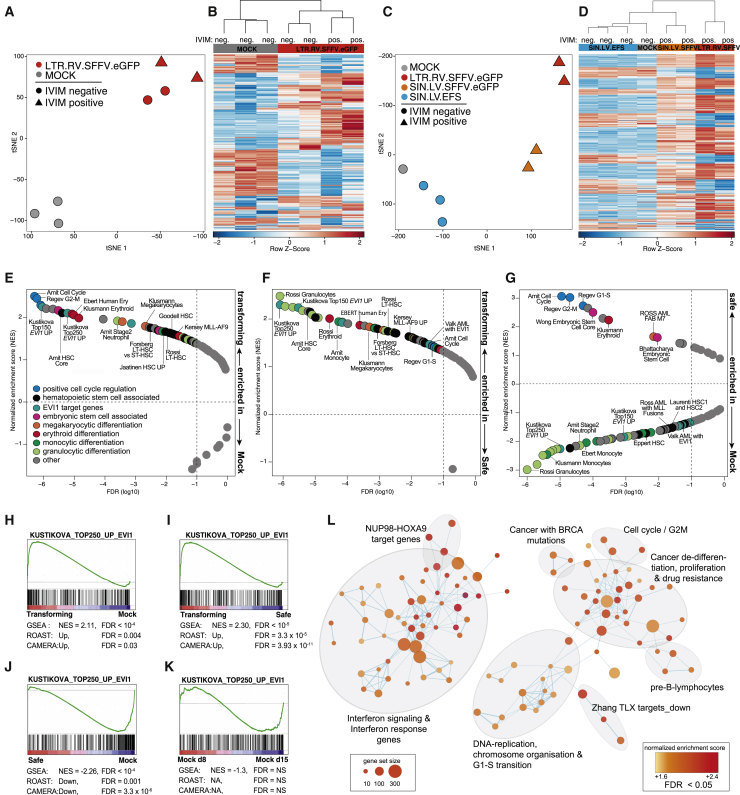
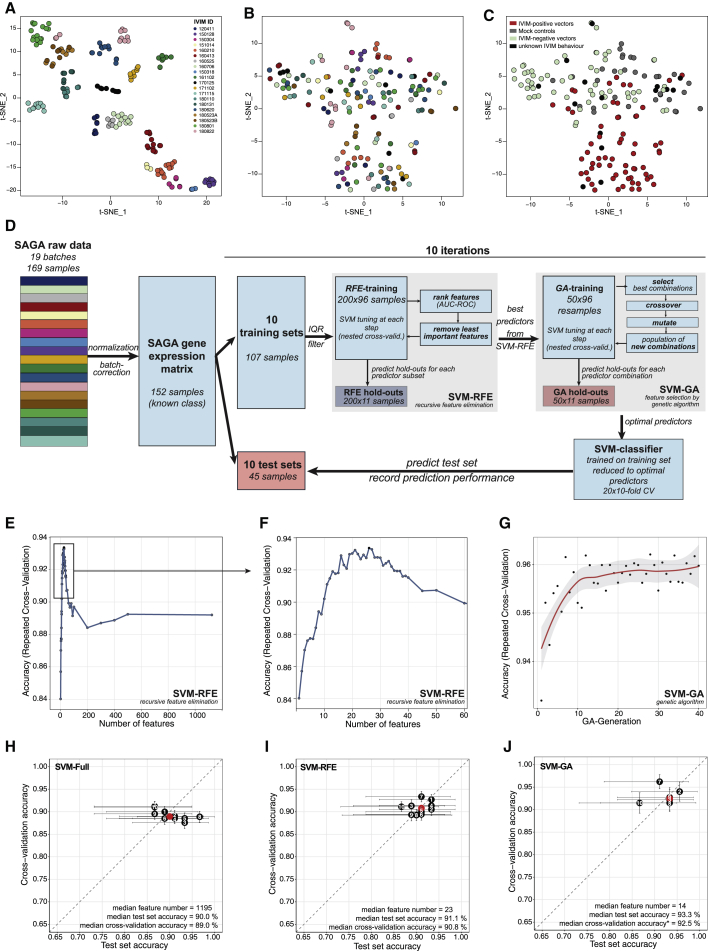
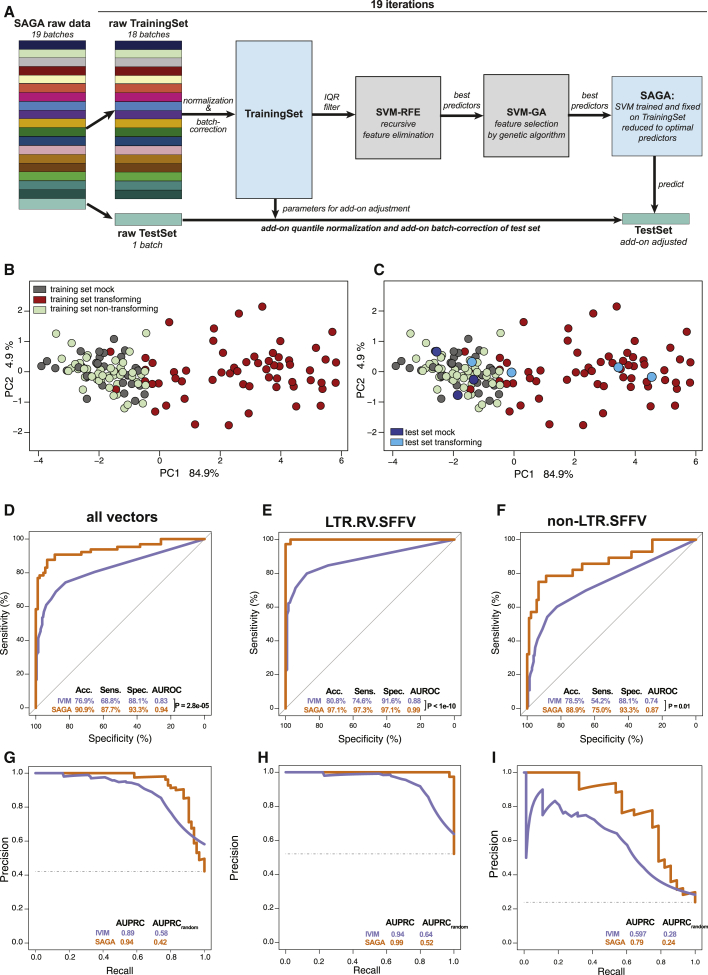
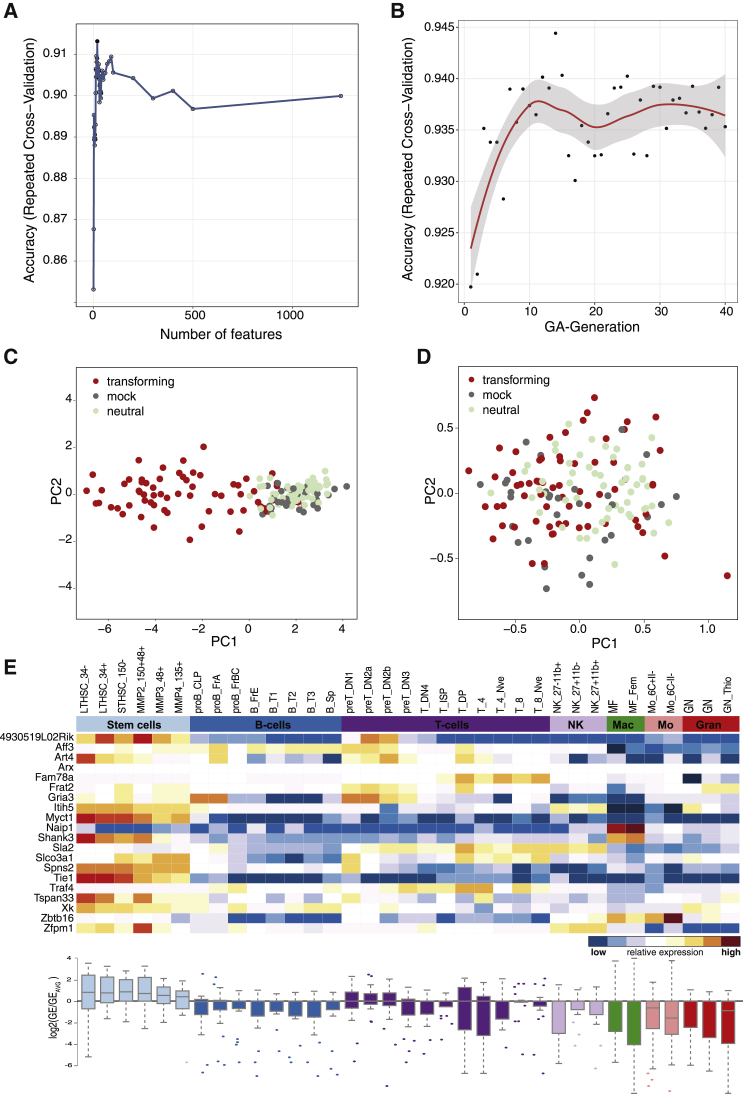
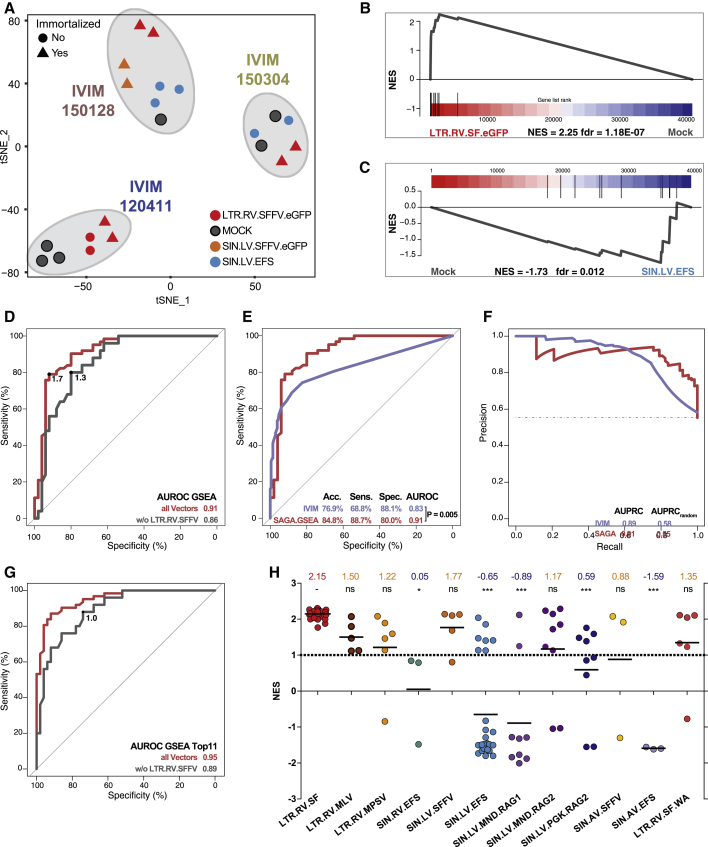
Hematopoietic stem cell gene therapy is emerging as a promising therapeutic strategy for many diseases of the blood and immune system. However, several individuals who underwent gene therapy in different trials developed hematological malignancies caused by insertional mutagenesis. Preclinical assessment of vector safety remains challenging because there are few reliable assays to screen for potential insertional mutagenesis effects in vitro. Here we demonstrate that genotoxic vectors induce a unique gene expression signature linked to stemness and oncogenesis in transduced murine hematopoietic stem and progenitor cells. Based on this finding, we developed the surrogate assay for genotoxicity assessment (SAGA). SAGA classifies integrating retroviral vectors using machine learning to detect this gene expression signature during the course of in vitro immortalization. On a set of benchmark vectors with known genotoxic potential, SAGA achieved an accuracy of 90.9%. SAGA is more robust and sensitive and faster than previous assays and reliably predicts a mutagenic risk for vectors that led to leukemic severe adverse events in clinical trials. Our work provides a fast and robust tool for preclinical risk assessment of gene therapy vectors, potentially paving the way for safer gene therapy trials.
Keywords: gene expression; gene therapy; genotoxicity; in vitro assay; insertional mutagenesis; integrating viral vectors; machine learning; preclinical risk assessment; safety assay gene therapy; support vector machine.
Copyright © 2021 The Authors. Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of interests A patent application has been filed under registration number EP3394286A1 (Analytical process for genotoxicity assessment).
Figures
References
-
- Hacein-Bey-Abina S., von Kalle C., Schmidt M., Le Deist F., Wulffraat N., McIntyre E., Radford I., Villeval J.-L., Fraser C.C., Cavazzana-Calvo M., Fischer A. A serious adverse event after successful gene therapy for X-linked severe combined immunodeficiency. N. Engl. J. Med. 2003;348:255–256. - PubMed
-
- Stein S., Ott M.G., Schultze-Strasser S., Jauch A., Burwinkel B., Kinner A., Schmidt M., Krämer A., Schwäble J., Glimm H., et al. Genomic instability and myelodysplasia with monosomy 7 consequent to EVI1 activation after gene therapy for chronic granulomatous disease. Nat. Med. 2010;16:198–204. - PubMed
-
- Braun C.J., Boztug K., Paruzynski A., Witzel M., Schwarzer A., Rothe M., Modlich U., Beier R., Gohring G., Steinemann D., et al. Gene Therapy for Wiskott-Aldrich Syndrome–Long-Term Efficacy and Genotoxicity. Sci. Transl. Med. 2014;6:227ra33. - PubMed
-
- Howe S.J., Mansour M.R., Schwarzwaelder K., Bartholomae C., Hubank M., Kempski H., Brugman M.H., Pike-Overzet K., Chatters S.J., de Ridder D., et al. Insertional mutagenesis combined with acquired somatic mutations causes leukemogenesis following gene therapy of SCID-X1 patients. J. Clin. Invest. 2008;118:3143–3150. - PMC - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
