

Mitophagy coordinates the mitochondrial unfolded protein response to attenuate inflammation-mediated myocardial injury
- PMID: 34174558
- PMCID: PMC8246635
- DOI: 10.1016/j.redox.2021.102049
Mitophagy coordinates the mitochondrial unfolded protein response to attenuate inflammation-mediated myocardial injury
Abstract
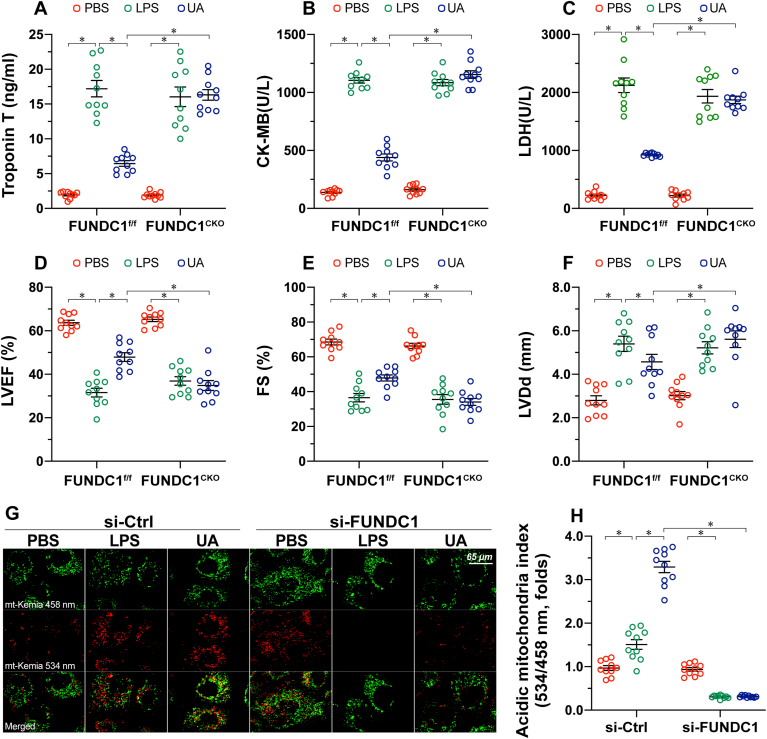
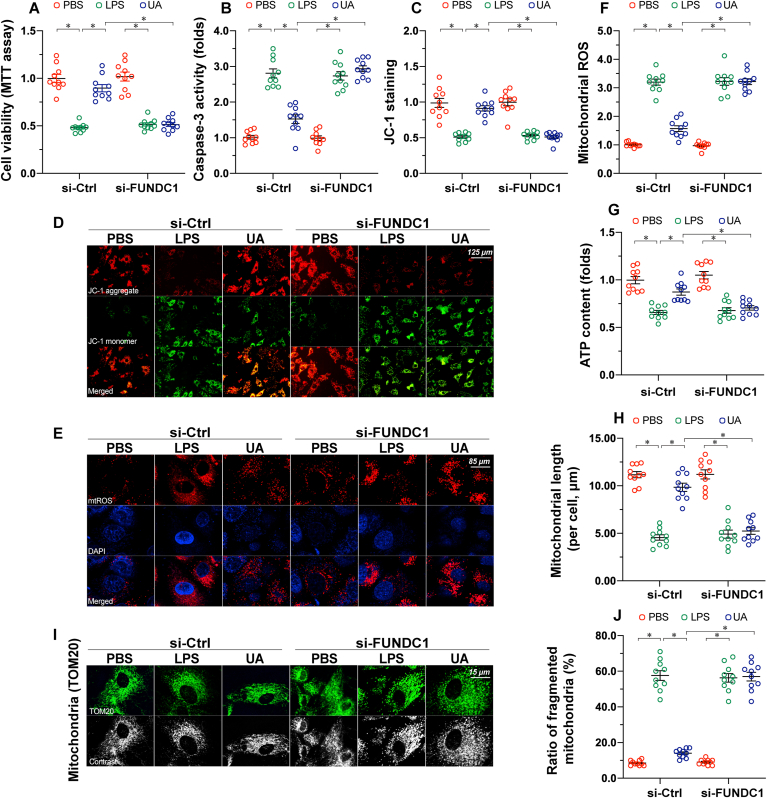
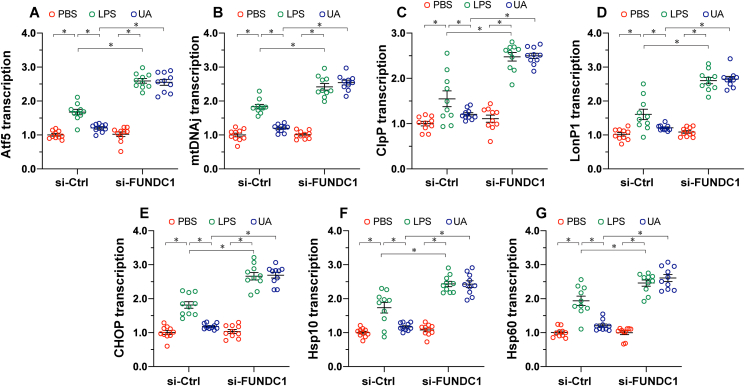
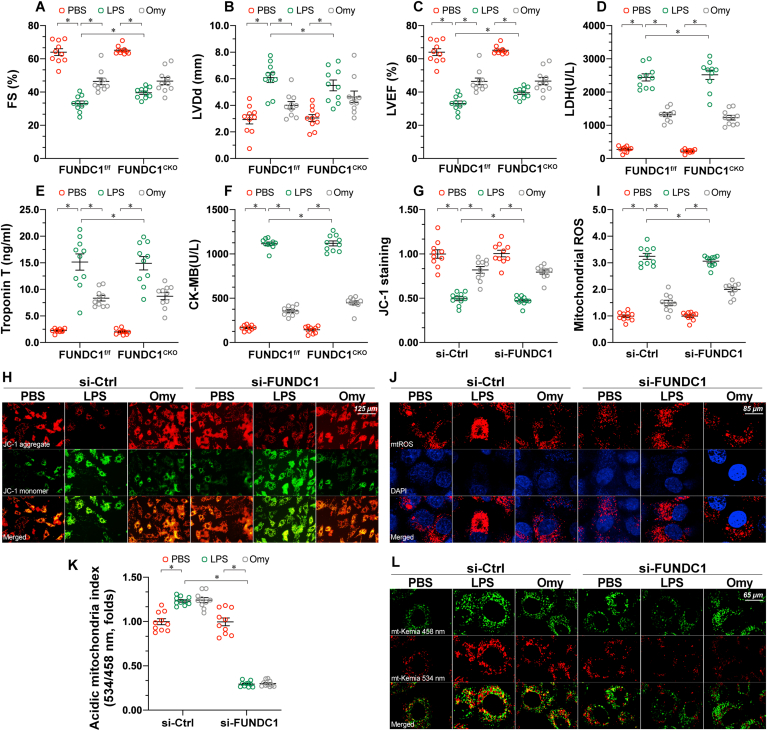
Mitochondrial dysfunction is a fundamental challenge in septic cardiomyopathy. Mitophagy and the mitochondrial unfolded protein response (UPRmt) are the predominant stress-responsive and protective mechanisms involved in repairing damaged mitochondria. Although mitochondrial homeostasis requires the coordinated actions of mitophagy and UPRmt, their molecular basis and interactive actions are poorly understood in sepsis-induced myocardial injury. Our investigations showed that lipopolysaccharide (LPS)-induced sepsis contributed to cardiac dysfunction and mitochondrial damage. Although both mitophagy and UPRmt were slightly activated by LPS in cardiomyocytes, their endogenous activation failed to prevent sepsis-mediated myocardial injury. However, administration of urolithin A, an inducer of mitophagy, obviously reduced sepsis-mediated cardiac depression by normalizing mitochondrial function. Interestingly, this beneficial action was undetectable in cardiomyocyte-specific FUNDC1 knockout (FUNDC1CKO) mice. Notably, supplementation with a mitophagy inducer had no impact on UPRmt, whereas genetic ablation of FUNDC1 significantly upregulated the expression of genes related to UPRmt in LPS-treated hearts. In contrast, enhancement of endogenous UPRmt through oligomycin administration reduced sepsis-mediated mitochondrial injury and myocardial dysfunction; this cardioprotective effect was imperceptible in FUNDC1CKO mice. Lastly, once UPRmt was inhibited, mitophagy-mediated protection of mitochondria and cardiomyocytes was partly blunted. Taken together, it is plausible that endogenous UPRmt and mitophagy are slightly activated by myocardial stress and they work together to sustain mitochondrial performance and cardiac function. Endogenous UPRmt, a downstream signal of mitophagy, played a compensatory role in maintaining mitochondrial homeostasis in the case of mitophagy inhibition. Although UPRmt activation had no negative impact on mitophagy, UPRmt inhibition compromised the partial cardioprotective actions of mitophagy. This study shows how mitophagy modulates UPRmt to attenuate inflammation-related myocardial injury and suggests the potential application of mitophagy and UPRmt targeting in the treatment of myocardial stress.
Keywords: FUN14 domain-containing 1; Inflammation; Mitochondrial unfolded protein response; Mitophagy; Septic cardiomyopathy.
Copyright © 2021 The Authors. Published by Elsevier B.V. All rights reserved.
Conflict of interest statement
All authors declare that they have no conflict of interest.
Figures
Similar articles
-
FUNDC1 activates the mitochondrial unfolded protein response to preserve mitochondrial quality control in cardiac ischemia/reperfusion injury.Cell Signal. 2022 Apr;92:110249. doi: 10.1016/j.cellsig.2022.110249. Epub 2022 Jan 17. Cell Signal. 2022. PMID: 35051611
-
BI-1 ameliorates myocardial injury by activating the mitochondrial unfolded protein response and FUNDC1-related mitophagy in cardiorenal syndrome type 3.Cell Signal. 2022 Mar;91:110218. doi: 10.1016/j.cellsig.2021.110218. Epub 2021 Dec 16. Cell Signal. 2022. PMID: 34921980
-
Exploiting Mitochondria by Triggering a Faulty Unfolded Protein Response Leads to Effective Cardioprotection.Int J Med Sci. 2025 Jan 1;22(1):188-196. doi: 10.7150/ijms.100523. eCollection 2025. Int J Med Sci. 2025. PMID: 39744160 Free PMC article.
-
Mitochondria Retrograde Signaling and the UPR mt: Where Are We in Mammals?Int J Mol Sci. 2015 Aug 6;16(8):18224-51. doi: 10.3390/ijms160818224. Int J Mol Sci. 2015. PMID: 26258774 Free PMC article. Review.
-
Enhancement of Mitochondrial Homeostasis: A Novel Approach to Attenuate Hypoxic Myocardial Injury.Int J Med Sci. 2024 Oct 28;21(15):2897-2911. doi: 10.7150/ijms.103986. eCollection 2024. Int J Med Sci. 2024. PMID: 39628681 Free PMC article. Review.
Cited by
-
The Mitochondrial Unfolded Protein Response: A Novel Protective Pathway Targeting Cardiomyocytes.Oxid Med Cell Longev. 2022 Sep 21;2022:6430342. doi: 10.1155/2022/6430342. eCollection 2022. Oxid Med Cell Longev. 2022. PMID: 36187338 Free PMC article. Review.
-
Insight into the mitochondrial unfolded protein response and cancer: opportunities and challenges.Cell Biosci. 2022 Feb 18;12(1):18. doi: 10.1186/s13578-022-00747-0. Cell Biosci. 2022. PMID: 35180892 Free PMC article. Review.
-
Xuebijing injection protects sepsis induced myocardial injury by mediating TLR4/NF-κB/IKKα and JAK2/STAT3 signaling pathways.Aging (Albany NY). 2023 Aug 30;15(16):8501-8517. doi: 10.18632/aging.204990. Epub 2023 Aug 30. Aging (Albany NY). 2023. PMID: 37650558 Free PMC article.
-
The Role of Mitochondrial Mutations in Chronification of Inflammation: Hypothesis and Overview of Own Data.Life (Basel). 2022 Jul 29;12(8):1153. doi: 10.3390/life12081153. Life (Basel). 2022. PMID: 36013333 Free PMC article.
-
SIRT3 and RORα are two prospective targets against mitophagy during simulated ischemia/reperfusion injury in H9c2 cells.Heliyon. 2024 May 8;10(10):e30568. doi: 10.1016/j.heliyon.2024.e30568. eCollection 2024 May 30. Heliyon. 2024. PMID: 38784556 Free PMC article.
References
-
- Hollenberg S.M., Singer M. Pathophysiology of sepsis-induced cardiomyopathy. Nat. Rev. Cardiol. 2021;18(6):424–434. - PubMed
-
- Vasques-Nóvoa F., Angélico-Gonçalves A., Bettencourt N., Leite-Moreira A.F., Roncon-Albuquerque R., Jr. Myocardial edema and remodeling: a link between acute myocarditis and septic cardiomyopathy? J. Am. Coll. Cardiol. 2020;75(12):1497–1498. - PubMed
-
- Sanfilippo F., Orde S., Oliveri F., Scolletta S., Astuto M. The challenging diagnosis of septic cardiomyopathy. Chest. 2019;156(3):635–636. - PubMed
-
- Zhong J., Tan Y., Lu J., Liu J., Xiao X., Zhu P., Chen S., Zheng S., Chen Y., Hu Y., Guo Z. Therapeutic contribution of melatonin to the treatment of septic cardiomyopathy: a novel mechanism linking Ripk3-modified mitochondrial performance and endoplasmic reticulum function. Redox Biol. 2019;26:101287. - PMC - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
