

LABRAT reveals association of alternative polyadenylation with transcript localization, RNA binding protein expression, transcription speed, and cancer survival
- PMID: 34174817
- PMCID: PMC8234626
- DOI: 10.1186/s12864-021-07781-1
LABRAT reveals association of alternative polyadenylation with transcript localization, RNA binding protein expression, transcription speed, and cancer survival
Abstract
Background: The sequence content of the 3' UTRs of many mRNA transcripts is regulated through alternative polyadenylation (APA). The study of this process using RNAseq data, though, has been historically challenging.
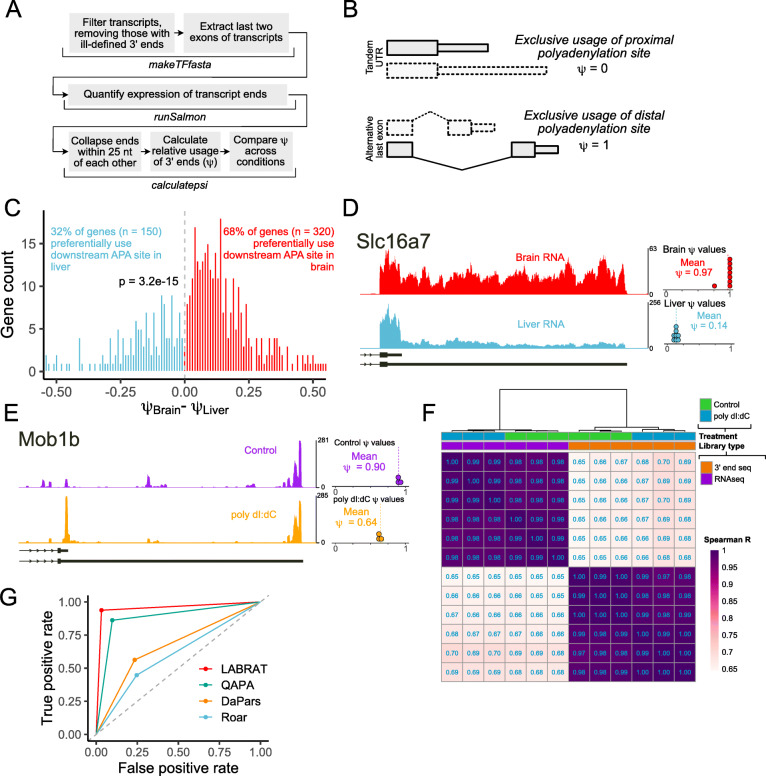
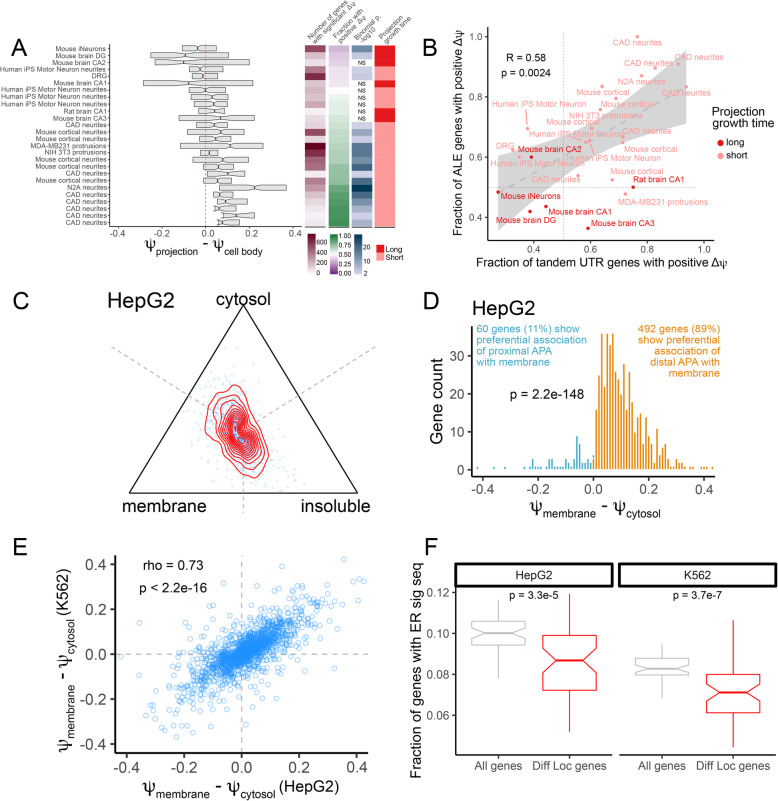
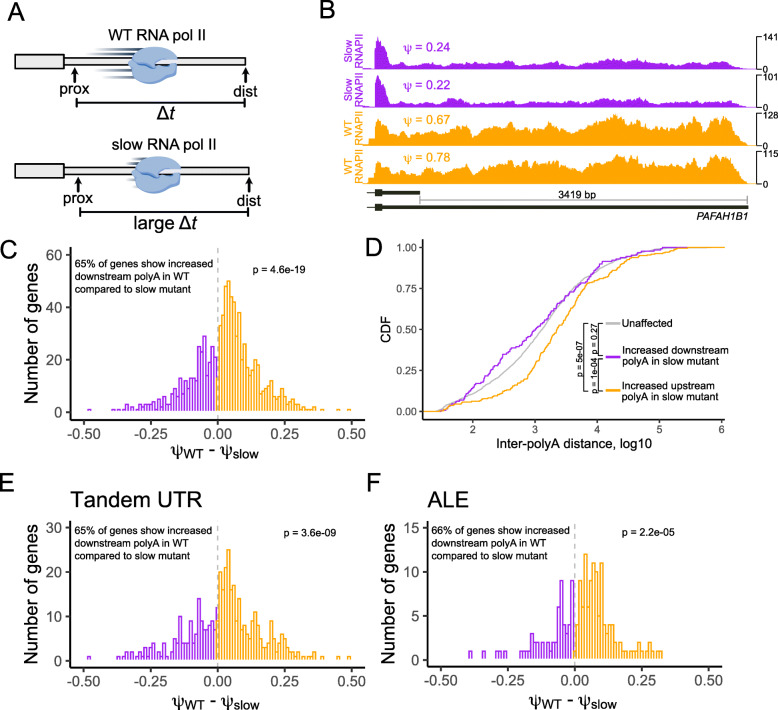
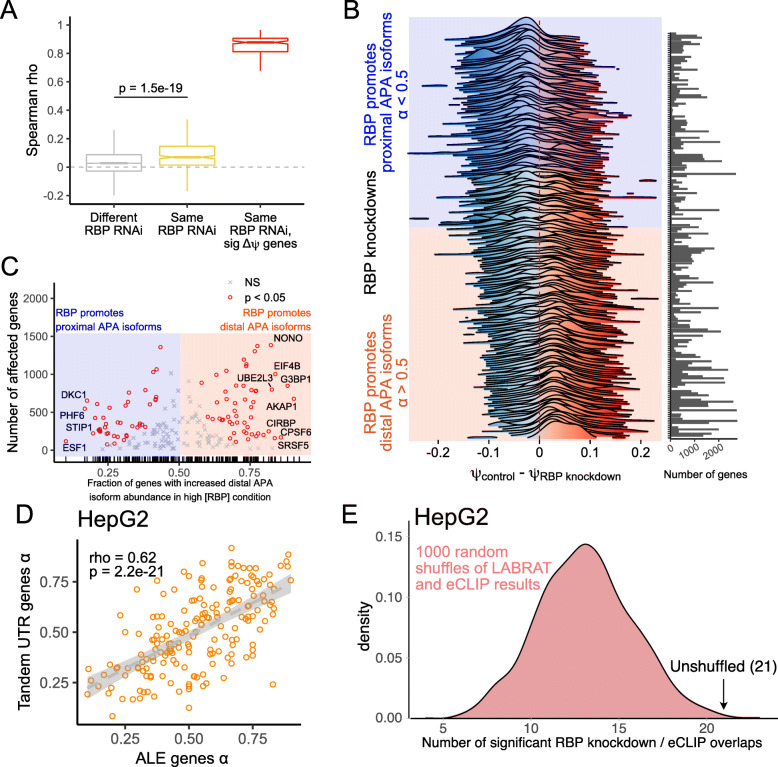
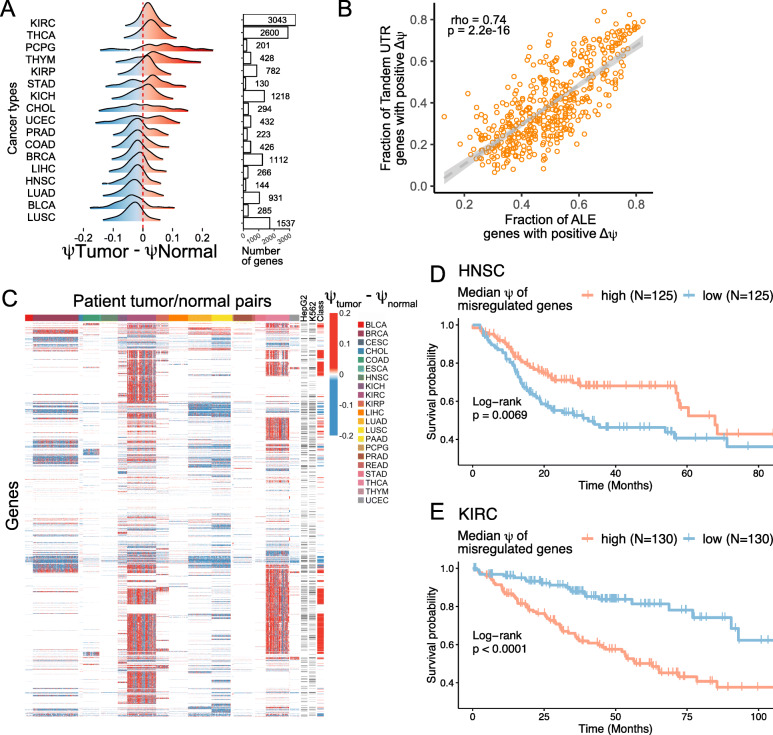
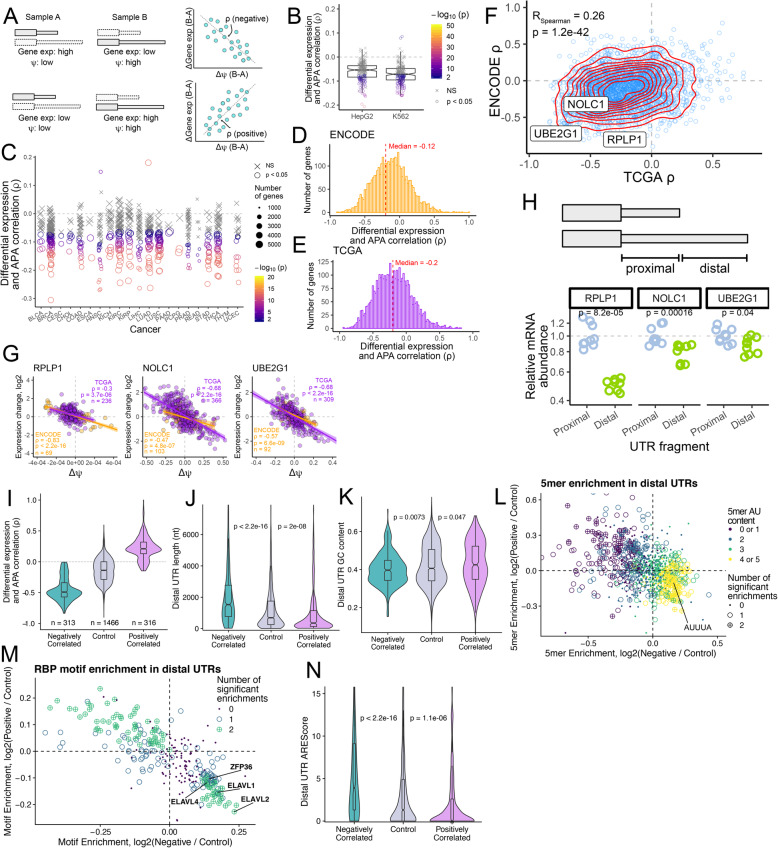
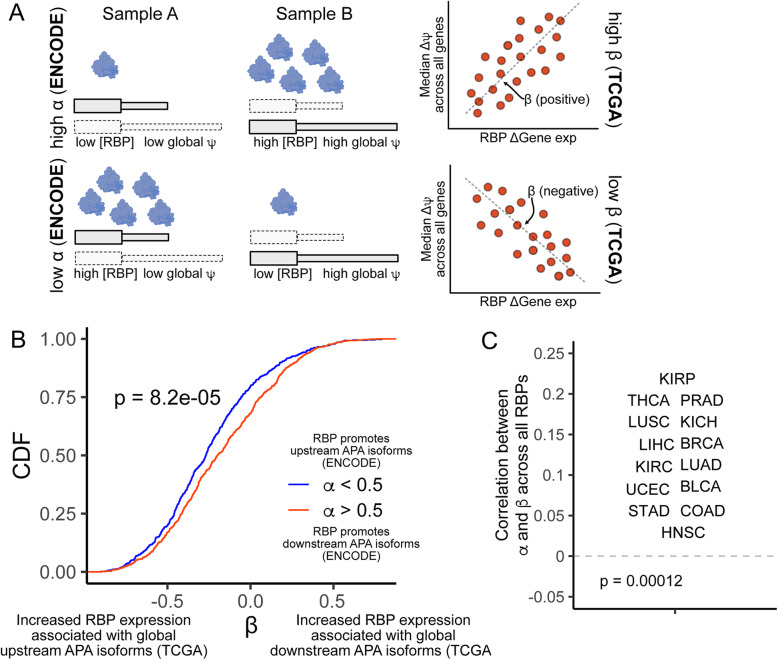
Results: To combat this problem, we developed LABRAT, an APA isoform quantification method. LABRAT takes advantage of newly developed transcriptome quantification techniques to accurately determine relative APA site usage and how it varies across conditions. Using LABRAT, we found consistent relationships between gene-distal APA and subcellular RNA localization in multiple cell types. We also observed connections between transcription speed and APA site choice as well as tumor-specific transcriptome-wide shifts in APA isoform abundance in hundreds of patient-derived tumor samples that were associated with patient prognosis. We investigated the effects of APA on transcript expression and found a weak overall relationship, although many individual genes showed strong correlations between relative APA isoform abundance and overall gene expression. We interrogated the roles of 191 RNA-binding proteins in the regulation of APA isoforms, finding that dozens promote broad, directional shifts in relative APA isoform abundance both in vitro and in patient-derived samples. Finally, we find that APA site shifts in the two classes of APA, tandem UTRs and alternative last exons, are strongly correlated across many contexts, suggesting that they are coregulated.
Conclusions: We conclude that LABRAT has the ability to accurately quantify APA isoform ratios from RNAseq data across a variety of sample types. Further, LABRAT is able to derive biologically meaningful insights that connect APA isoform regulation to cellular and molecular phenotypes.
Conflict of interest statement
None.
Figures
References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
