

Biallelic Mutations in DNAJB11 are Associated with Prenatal Polycystic Kidney Disease in a Turkish Family
- PMID: 34177435
- PMCID: PMC8216001
- DOI: 10.1159/000513611
Biallelic Mutations in DNAJB11 are Associated with Prenatal Polycystic Kidney Disease in a Turkish Family
Abstract
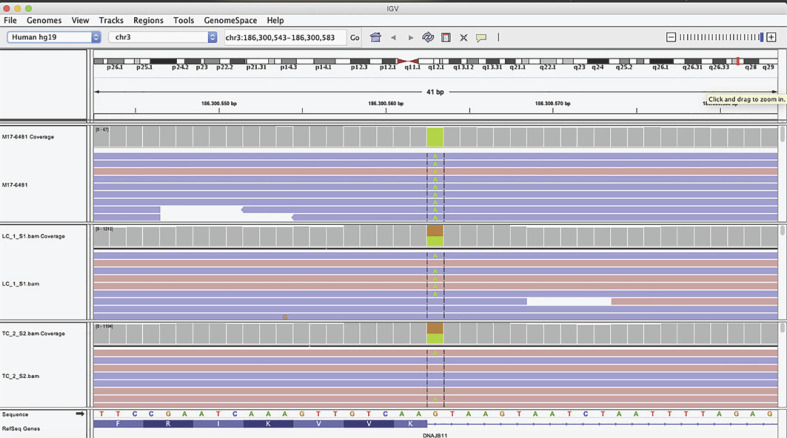
Polycystic kidney disease (PKD) is a life-threatening condition resulting in end-stage renal disease. Two major forms of PKD are defined according to the inheritance pattern. Autosomal dominant PKD (ADPKD) is characterized by renal cysts, where nearly half of the patients suffers from renal failure in the 7th decade of life. Autosomal recessive PKD (ARPKD) is a rarer and more severe form presenting in childhood. Whole-exome sequencing (WES) analyses was performed to investigate molecular causes of the disease in the fetus. In this study, we present 2 fetuses prenatally diagnosed with PKD in a consanguineous family. WES analysis of the second fetus revealed a homozygous variant (c.740+1G>A) in DNAJB11 which is related to ADPKD. This study reveals that DNAJB11 biallelic mutations may cause an antenatal severe form of ARPKD and contributes to understanding the DNAJB11-related ADPKD phenotype. The possibility of ARPKD due to biallelic mutations in ADPKD genes should be considered in genetic counseling.
Keywords: DNAJB11; Genetic counseling; Nephrology; Polycystic kidney disease; Whole-exome sequencing.
Copyright © 2021 by S. Karger AG, Basel.
Conflict of interest statement
The authors have no conflicts of interest to declare.
Figures


References
-
- Al-Hamed MH, Alsahan N, Rice SJ, Edwards N, Nooreddeen E, Alotaibi M, et al. Bialleleic PKD1 mutations underlie early-onset autosomal dominant polycystic kidney disease in Saudi Arabian families. Pediatr Nephrol. 2019;34((9)):1615–23. - PubMed
-
- Alzarka B, Morizono H, Bollman JW, Kim D, Guay-Woodford LM. Design and Implementation of the Hepatorenal Fibrocystic Disease Core Center Clinical Database: A Centralized Resource for Characterizing Autosomal Recessive Polycystic Kidney Disease and Other Hepatorenal Fibrocystic Diseases. Front Pediatr. 2017;5:80. - PMC - PubMed
Publication types
LinkOut - more resources
Full Text Sources
