

From Genome to Drugs: New Approaches in Antimicrobial Discovery
- PMID: 34177572
- PMCID: PMC8219968
- DOI: 10.3389/fphar.2021.647060
From Genome to Drugs: New Approaches in Antimicrobial Discovery
Abstract
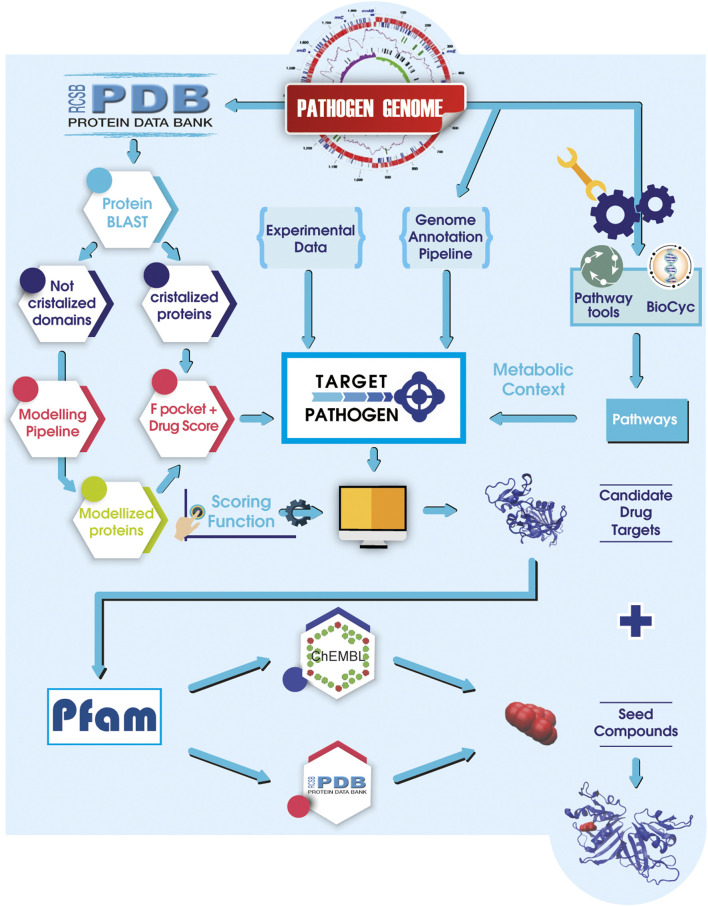
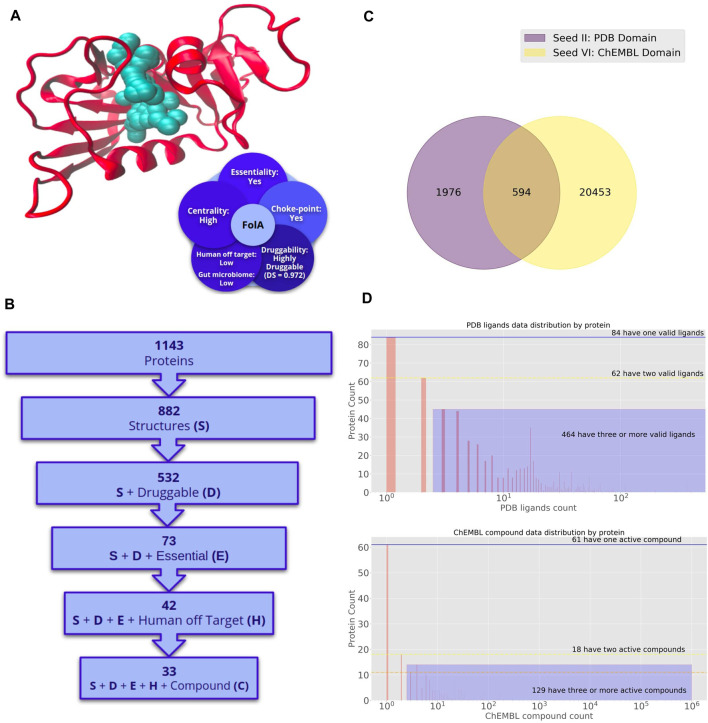
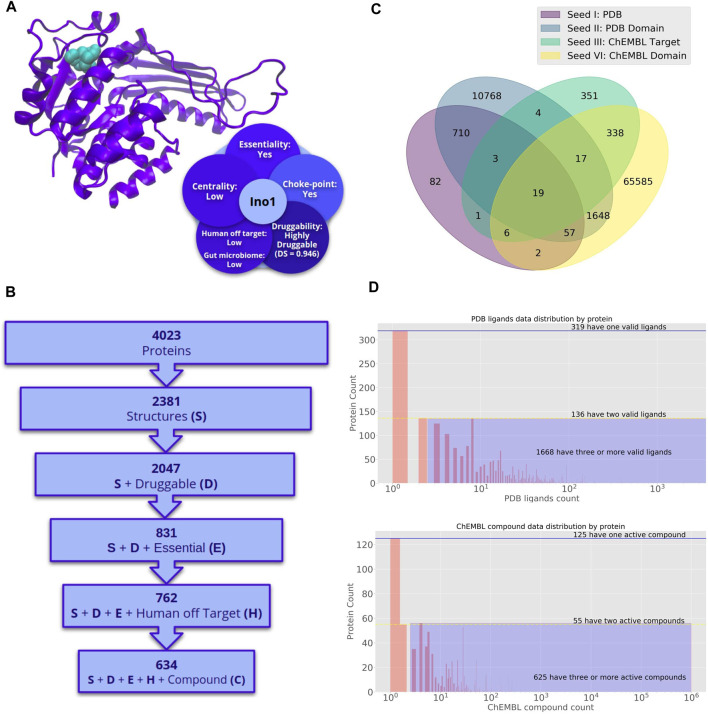
Decades of successful use of antibiotics is currently challenged by the emergence of increasingly resistant bacterial strains. Novel drugs are urgently required but, in a scenario where private investment in the development of new antimicrobials is declining, efforts to combat drug-resistant infections become a worldwide public health problem. Reasons behind unsuccessful new antimicrobial development projects range from inadequate selection of the molecular targets to a lack of innovation. In this context, increasingly available omics data for multiple pathogens has created new drug discovery and development opportunities to fight infectious diseases. Identification of an appropriate molecular target is currently accepted as a critical step of the drug discovery process. Here, we review how diverse layers of multi-omics data in conjunction with structural/functional analysis and systems biology can be used to prioritize the best candidate proteins. Once the target is selected, virtual screening can be used as a robust methodology to explore molecular scaffolds that could act as inhibitors, guiding the development of new drug lead compounds. This review focuses on how the advent of omics and the development and application of bioinformatics strategies conduct a "big-data era" that improves target selection and lead compound identification in a cost-effective and shortened timeline.
Keywords: drug discovery; drug target; metabolic reconstruction; structural modeling; target prioritization; virtual screening.
Copyright © 2021 Serral, Castello, Sosa, Pardo, Palumbo, Modenutti, Palomino, Lazarowski, Auzmendi, Ramos, Nicolás, Turjanski, Martí and Fernández Do Porto.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures
Similar articles
-
Editorial: Current status and perspective on drug targets in tubercle bacilli and drug design of antituberculous agents based on structure-activity relationship.Curr Pharm Des. 2014;20(27):4305-6. doi: 10.2174/1381612819666131118203915. Curr Pharm Des. 2014. PMID: 24245755
-
[Development of antituberculous drugs: current status and future prospects].Kekkaku. 2006 Dec;81(12):753-74. Kekkaku. 2006. PMID: 17240921 Review. Japanese.
-
Antimicrobial Compounds from Microorganisms.Antibiotics (Basel). 2022 Feb 22;11(3):285. doi: 10.3390/antibiotics11030285. Antibiotics (Basel). 2022. PMID: 35326749 Free PMC article. Review.
-
Omics of antimicrobials and antimicrobial resistance.Expert Opin Drug Discov. 2019 May;14(5):455-468. doi: 10.1080/17460441.2019.1588880. Epub 2019 Mar 19. Expert Opin Drug Discov. 2019. PMID: 30884978 Review.
-
Insights into Integrated Lead Generation and Target Identification in Malaria and Tuberculosis Drug Discovery.Acc Chem Res. 2017 Jul 18;50(7):1606-1616. doi: 10.1021/acs.accounts.6b00631. Epub 2017 Jun 21. Acc Chem Res. 2017. PMID: 28636311 Free PMC article.
Cited by
-
Computer-Based Identification of Potential Druggable Targets in Multidrug-Resistant Acinetobacter baumannii: A Combined In Silico, In Vitro and In Vivo Study.Microorganisms. 2022 Oct 5;10(10):1973. doi: 10.3390/microorganisms10101973. Microorganisms. 2022. PMID: 36296249 Free PMC article.
-
Repurposing Selamectin as an Antimicrobial Drug against Hospital-Acquired Staphylococcus aureus Infections.Microorganisms. 2023 Sep 6;11(9):2242. doi: 10.3390/microorganisms11092242. Microorganisms. 2023. PMID: 37764086 Free PMC article.
-
In vitro antimicrobial activity and resistance mechanisms of the new generation tetracycline agents, eravacycline, omadacycline, and tigecycline against clinical Staphylococcus aureus isolates.Front Microbiol. 2022 Nov 22;13:1043736. doi: 10.3389/fmicb.2022.1043736. eCollection 2022. Front Microbiol. 2022. PMID: 36483205 Free PMC article.
-
Unrealized targets in the discovery of antibiotics for Gram-negative bacterial infections.Nat Rev Drug Discov. 2023 Dec;22(12):957-975. doi: 10.1038/s41573-023-00791-6. Epub 2023 Oct 13. Nat Rev Drug Discov. 2023. PMID: 37833553 Review.
-
Synthesis of Silver Nanoparticles and Evaluation of Antimicrobial Activity Using the Aqueous Extract of Pterodon emarginatus Seeds.Cureus. 2024 Dec 25;16(12):e76382. doi: 10.7759/cureus.76382. eCollection 2024 Dec. Cureus. 2024. PMID: 39722661 Free PMC article.
References
-
- Sussman J. L., Lin D., Jiang J., Manning N. O., Prilusky J., Ritter O., et al. (1998). Protein Data Bank (PDB): database of three-dimensional structural information of biological macromolecules. Acta Crystallogr. D Biol. Crystallogr. 54, 1078–1084. - PubMed
-
- Andrade C. H., Pasqualoto K. F. M., Zaim M. H., Ferreira E. I. (2008). Abordagem racional no planejamento de novos tuberculostáticos: inibidores da InhA, enoil-ACP redutase Do M. tuberculosis . Rev. Bras. Cienc. Farm. 44, 167–179. 10.1590/s1516-93322008000200002 - DOI
Publication types
LinkOut - more resources
Full Text Sources